Metformin enhances the cytotoxic effect of nilotinib and overcomes nilotinib resistance in chronic myeloid leukemia cells
Article information

Abstract
Background/Aims
Nilotinib is used for treating patients with imatinib-sensitive or -resistant chronic myeloid leukemia (CML); however, nilotinib-resistant cases have been observed in recent years. In addition, a considerable number of patients receiving nilotinib developed diabetes. Metformin is a front-line drug for the treatment of type 2 diabetes, and several studies have shown that diabetes patients treated with metformin have reduced incidence of cancer. This study aimed to define the effect of metformin on CML cells to determine whether metformin overcomes nilotinib resistance, and to identify novel targets for the treatment of nilotinib resistance.
Methods
We observed the effects of metformin and nilotinib on K562 and KU812 human CML cell lines. Nilotinib-resistant CML cell lines were generated by exposing cells to gradually increasing doses of nilotinib. Then, we investigated the driving force that makes resistance to nilotinib and the effect of metformin on the driving force.
Results
Sub-toxic doses of metformin enhanced nilotinib efficacy by reducing Bcl-xL expression, which induces apoptosis in CML cells. Next, we generated nilotinib-resistant K562 and KU812 cell lines that overexpressed the c-Jun N-terminal kinase (JNK) gene. JNK silencing by a JNK inhibitor restored sensitivity to nilotinib. Furthermore, metformin was effective in decreasing phosphorylated JNK levels, restoring nilotinib sensitivity. Combined treatment with nilotinib and metformin was more effective than combined treatment with nilotinib and a JNK inhibitor in terms of cell proliferation inhibition.
Conclusions
This study suggested that combination therapy with metformin and nilotinib may have clinical benefits of enhancing antileukemia efficacy and overcoming resistance to nilotinib.
INTRODUCTION
Chronic myeloid leukemia (CML) is characterized by the presence of the Philadelphia chromosome with a reciprocal translocation between chromosomes 9 and 22 [1,2], and this translocation generates the Bcr/Abl fusion gene. The Bcr/Abl fusion gene produces a tyrosine kinase that drives oncogenesis by activating downstream signals, such as RAS/RAF/MEK/ERK, and prevents apoptosis [3].
Nilotinib is a second-generation tyrosine kinase inhibitor (TKI) that competes with ATP, resulting in selective inhibition of Bcr/Abl activity [4]. Nilotinib has the mechanism of action similar to that of imatinib mesylate, a first-generation TKI; however, nilotinib is more potent than imatinib in treating imatinib-sensitive or -resistant CML patients. Nonetheless, a considerable number of patients receiving nilotinib have presented new-onset diabetes and impaired glucose metabolism with increased blood glucose level (hyperglycemia) [5]. This “hyperglycemic” effect of nilotinib makes it unacceptable to patients with diabetes. More importantly, many cases of resistance to nilotinib, as in imatinib [6], have recently been reported as a major concern [7].
Metformin is a first-line drug widely used for the treatment of type 2 diabetes mellitus; it blocks hepatic glucose production through an LKB1/AMPK-mediated mechanism. Lately, several studies have reported that metformin not only inhibits cell proliferation in breast, gastric, ovarian, and small cell lung cancers [8-11] but also potentiates the cytotoxic effect of some TKIs in bladder, colorectal, and non-small cell lung cancer [12-14]. In addition, it is also known to reverse multidrug resistance by regulating cancer stem cell population or signaling [15-17]. However, the effects of metformin on CML have not been elucidated.
This study aimed to define the effect of metformin on nilotinib resistance in CML cells and to identify novel targets for treating nilotinib resistance.
METHODS
Cell culture
Human CML K562 cells were purchased from the Korea Cell Line Bank (Seoul, Korea), and KU812 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in RPMI 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum and antibiotic-antimycotic solution (Life Technologies, Carlsbad, CA, USA). The cells were cultured in a humidified incubator at 37°C with 5% CO2.
Drugs and antibodies
Nilotinib was purchased from Cayman (Ann Arbor, MI, USA) and was dissolved in dimethyl sulfoxide. Metformin hydrochloride was purchased from Wako (Richmond, VA, USA) and was dissolved in distilled water. Rabbit antibodies against c-Jun N-terminal kinase (JNK), phosphor-JNK, ERK, phosphor-ERK, p38, phosphor-p38, BCR-ABL, phosphor-BCR-ABL, caspase 3, caspase 8, PARP, and Bid, as well as mouse antibody against caspase 9 were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Mouse antibody against Bcl-2 and rabbit antibody against Bcl-xL were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-actin was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell proliferation assay
For the cell proliferation assay, we performed an EZ-Cytox assay. Aliquots of cells (8 × 103) were plated into 96-well plates (200 μL growth medium per well) and then treated with metformin or nilotinib at various concentrations. After 48 hours of incubation, 20 μL of the Kit reagent (EZ-Cytox Kit, DoGEN, Seoul, Korea) was added to each well. Next, the cells were incubated at 37°C for 3 hours, and the absorbance was measured at 450 nm by a microplate reader.
Cell survival assay
For the trypan blue exclusion assay, centrifuged cells were resuspended in 1 mL of phosphate-buffered saline solution (PBS) and then mixed with a 0.4% trypan blue solution at a 1:1 ratio. The samples were incubated at 25°C for 5 minutes and visualized by light microscopy. At least 200 cells were counted for each survival determination.
Annexin V binding assay
Cells were harvested and resuspended in 100 μL of binding buffer provided in the Annexin V-FITC apoptosis detection kit (BioBud, Seoul, Korea). Cells were mixed with 1.25 μL Annexin V-FITC reagent and 10 μL propidium iodide (PI) and incubated for 15 minutes at room temperature in the dark. The staining was terminated by addition of binding buffer and the cells were immediately analyzed by flow cytometry (Beckman Coulter, Atlanta, GA, USA).
Western blotting analysis
After various treatments, the cells were washed with PBS before RIPA buffer (Sigma-Aldrich) was added to the cells. A protease and phosphatase inhibitor cocktail (Sigma-Aldrich) was added to the RIPA buffer before lysing the cells. Protein concentrations were measured with a BCA protein assay reagent (Thermo Scientific, Waltham, MA, USA). Equal amounts of protein were separated by 10% to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then electro-transferred to a nitrocellulose membrane (Amersham, Arlington Heights, IL, USA). The membrane was blocked with 5% skim milk in Tris buffered saline (TBS)-Tween20 for 2 hours and incubated with primary antibodies overnight at 4°C. The membrane was then incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G as a secondary antibody. Immunoreactive proteins were visualized using a chemiluminescence reagent (DoGen).
DAPI nuclear staining
Cells were seeded on poly-L-lysine (Sigma-Aldrich) pre-coated cover slips on 12-well plates. After incubation for 24 hours, the cells were treated with 10 nM nilotinib for 48 hours. Next, the cells were harvested, washed with PBS, and fixed with 3.7% formaldehyde at room temperature for 15 minutes. The cells were then stained with DAPI (Invitrogen, Carlsbad, CA, USA) solution at 37°C for 10 minutes in the dark. The cover slips were mounted onto glass microscope slides and examined by fluorescence microscopy (Carl Zeiss, Oberkochen, Germany).
Generation of nilotinib-resistant CML cells
Cells were maintained in suspension cultures and gradually exposed to increasing concentrations of nilotinib, starting with a concentration of 1 nM and ending with a final dose of 10 nM nilotinib. The parental, sensitive cell lines were maintained without nilotinib to be used as controls.
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was carried out using the GraphPad Prism version 5 software (GraphPad Software, San Diego, CA, USA). The results were expressed as mean with standard error of the mean and evaluated using a paired Student’s t test. The results were considered statistically significant at p < 0.05.
RESULTS
Effect of metformin, nilotinib, and their combination on the proliferation and apoptosis of CML cells
To investigate the anti-proliferative efficacy of metformin and nilotinib, cells were treated as described in the Methods section. As shown in Fig. 1A, both metformin and nilotinib induced cytotoxicity in a dose-dependent manner. However, metformin alone did not significantly affect cell proliferation (Fig. 1A). Next, we investigated the effect of combined treatment with metformin and nilotinib in CML cell lines. First, we observed the effect of 5 mM metformin in combination with 5 nM nilotinib on CML cell morphology under a light microscope. After the combined treatment, cell adhesion to each other significantly decreased in comparison to control or nilotinib alone-treated cells (Fig. 1B). We further examined whether combined treatment with metformin and nilotinib enhances cell death/apoptosis compared to metformin or nilotinib alone in CML cell lines. After combined treatment for 48 hours, the cells were stained with trypan blue. The results showed that the combined treatment induced 40% to 50% cell death (Fig. 1C). To clarify the apoptosis mechanism, we performed a western blotting assay with anti-PARP; anti-caspase 3, 8, and 9; anti-Bcl-xL; anti-Bcl2; and anti-Bid. The results showed that combination treatment induced activation of caspase 3, 8, and 9, which led to an increase in PARP cleavage in K562 cells. Additionally, decreased expression of Bcl-xL (anti-apoptotic protein) was majorly responsible for induction of apoptosis after the treatment (Fig. 1D). Furthermore, when the cells were stained with annexin V and PI, the early apoptotic cell percentage after combination treatment was almost eight-fold higher than that after control treatment (Fig. 1E). Collectively, combination therapy with metformin and nilotinib enhanced apoptosis mediated by an increase in caspase activation by decreasing Bcl-xL expression in nilotinib-sensitive CML cells.

Effects of metformin, nilotinib, and their combination on the proliferation and apoptosis of chronic myeloid leukemia cell lines. (A) Proliferation of K562 and KU812 cells after treatment with various concentrations of metformin (0 to 10 mM) or nilotinib (0 to 50 nM) for 48 hours. (B) Morphological changes (arrows) in KU812 cells were measured by microscopy after treatment with 5 mM metformin and 5 nM nilotinib for 48 hours. Scale bar: 20 µm. (C) Trypan blue exclusion assay was performed to analyze cell death after drug treatment. (D) Western blotting analysis of co-treatment with metformin and nilotinib in K562 cells. (E) Apoptosis analysis by flow cytometry after staining with annexin V and propidium iodide in K562 cells. Error bars represent standard error of the mean from three separate experiments. ap < 0.05, bp < 0.01, respectively.
Generation of nilotinib-resistant cell lines
To simulate nilotinib resistance, Philadelphia chromosome-positive cell lines (K562 and KU812) were cultured in the presence of increasing concentrations of nilotinib. After approximately 6 months, the cell lines continued to survive in the presence of 10 nM nilotinib. Almost completely nilotinib-resistant cells were able to grow in the presence of nilotinib at concentrations of up to 10 nM; however, parental cell lines could not grow at this concentration (Fig. 2A). The IC50 of the nilotinib-resistant K562R and KU812R cells to nilotinib was 120.7 and 151.5 nM, respectively, which was six times higher than that of the corresponding parental cell lines (22.86 and 25.85 nM, respectively). Next, the cells were stained with DAPI after being exposed to nilotinib for 48 hours. The result indicated that nilotinib-resistant cells were spherical and had homogeneous nuclei, whereas nilotinib-sensitive cells showed apoptotic bodies (Fig. 2B). The survival of sensitive and resistant cells was measured using trypan blue exclusion assay. The survival of nilotinib-sensitive cells, but not nilotinib-resistant cells, was decreased in a dose-dependent manner by treatment with up to 10 nM nilotinib (Fig. 2C). Lastly, the effect of increasing concentrations of nilotinib on Bcr/Abl phosphorylation and apoptosis was evaluated by western blotting. Phosphorylated Bcr/Abl level was maintained in nilotinib-resistant cells, but not in nilotinib-sensitive cells, after treatment with 10 nM nilotinib. Similar results were observed for PARP, caspase 3, and Bcl-xL (Fig. 2D).
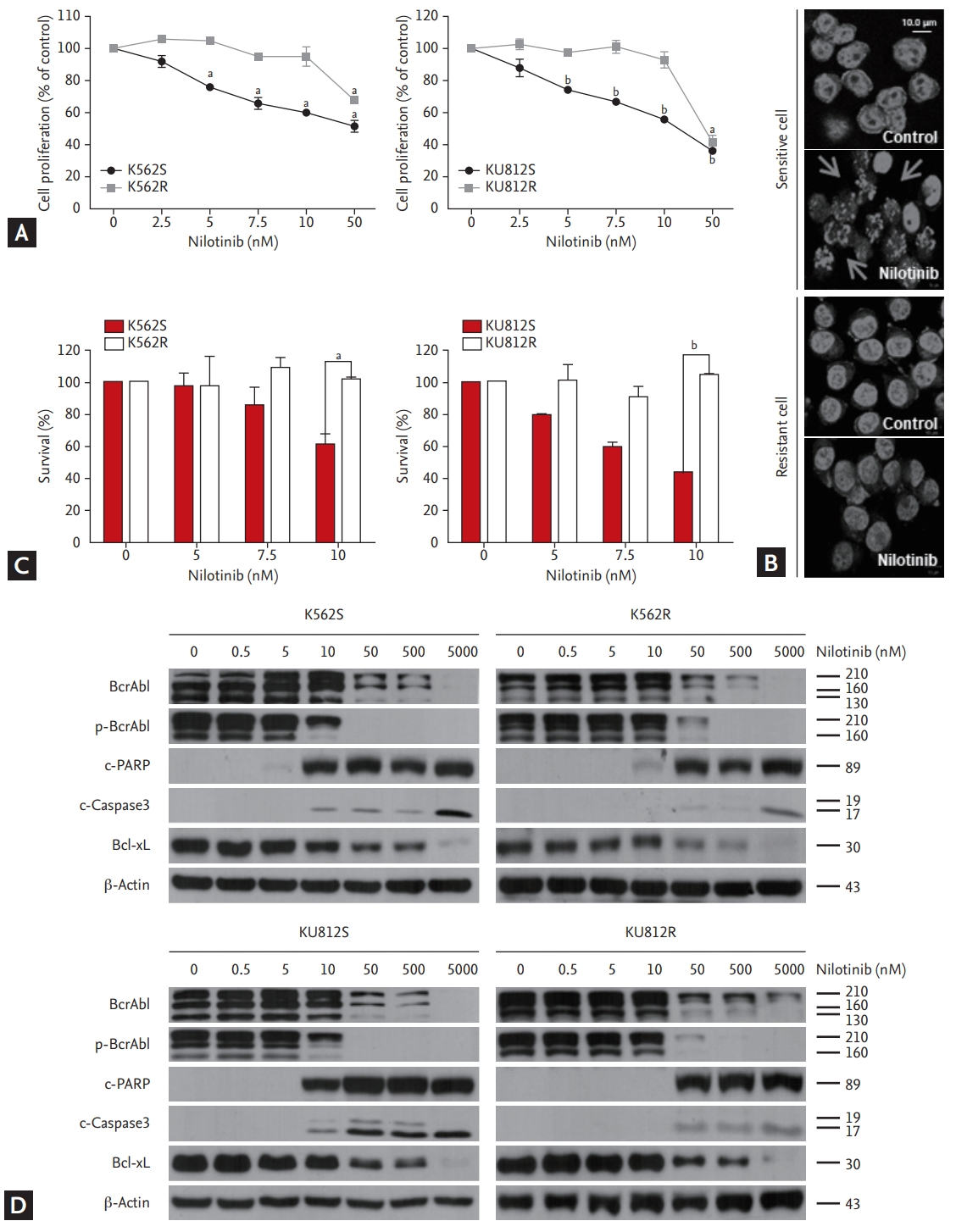
Generation of nilotinib-resistant cell lines. (A) EZ-Cytox assay for evaluation of cell proliferation in response to nilotinib alone in nilotinib-sensitive ‘S’ and nilotinib-resistant ‘R’ cells. At least three independent EZ-Cytox assays were performed using triplicate samples. (B) Morphological changes in K562 cells were measured by fluorescent m icroscopy after DAPI staining. Arrows show signs of nuclear shrinkage and chromatin condensation. (C) Trypan blue exclusion assay for determining the generation of nilotinib-resistant cells. (D) Western blotting analysis of cells after treatment with various concentrations of nilotinib for 48 hours. Bcr/Abl, phosphor-Bcr/Abl, PARP, caspase 3, and Bcl-xL were detected by immunoblotting. Actin was included as a loading control. ap < 0.01, bp < 0.001, respectively.
JNK overexpression and reversal of nilotinib resistance through inhibition of phosphor-JNK by metformin in nilotinib-resistant cells
To investigate whether nilotinib-resistant cell lines were associated with MAP kinase expression, western blotting was performed using anti-JNK, anti-ERK, and anti-p38 antibodies. The results indicated an overexpression of phosphor-JNK expression, but no significant differences in ERK and p38 expression were detected between nilotinib-sensitive and nilotinib-resistant cells (Fig. 3A). To confirm whether nilotinib resistance can be reversed by targeting the JNK gene, nilotinib-resistant cells were treated concomitantly with a JNK inhibitor (SP600125) and nilotinib. The JNK inhibitor (20 μM) together with increasing concentrations of nilotinib decreased the proliferation of nilotinib-resistant cells in a dose-dependent manner (Fig. 3B). Next, we observed whether metformin decreases JNK phosphorylation. The result indicated that metformin downregulated JNK phosphorylation in a dose-dependent manner in both nilotinib-sensitive and nilotinib-resistant cell lines (Fig. 3C). However, the treatment resulted in a significant reduction of JNK expression in nilotinib-resistant cells, compared to that in nilotinib-sensitive cells. To confirm whether combined treatment with metformin and nilotinib can restore nilotinib sensitivity by targeting the JNK protein, the EZ-Cytox assay was performed. Combined treatment with metformin (10 mM) and increasing concentrations of nilotinib decreased the proliferation of nilotinib-resistant cells in a dose-dependent manner (Fig. 3D). The cell growth inhibition effect of combined treatment with nilotinib and metformin was more potent than that of combined treatment with nilotinib and JNK inhibitor, as shown in Fig. 3B.
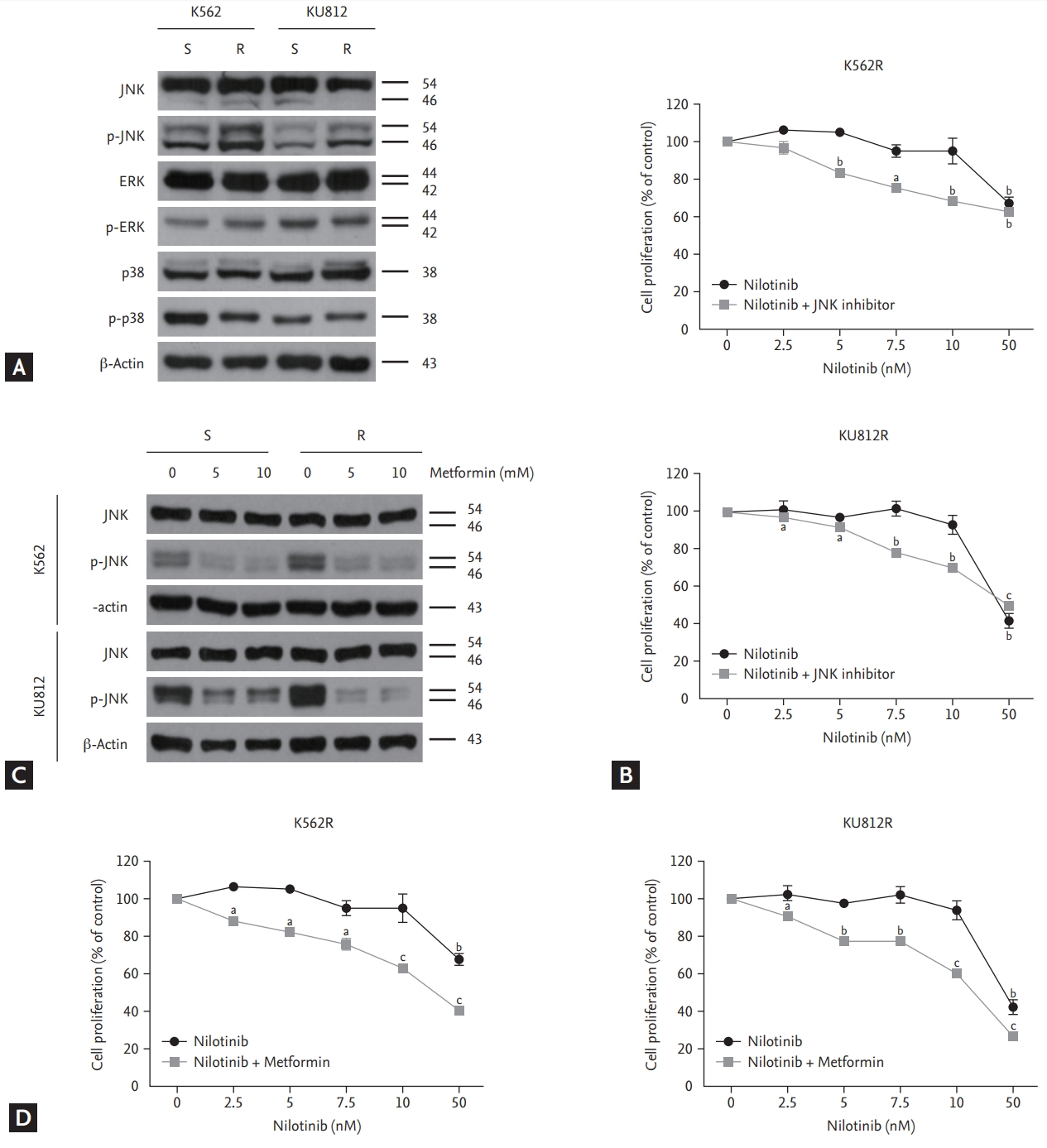
c-Jun N-terminal kinase (JNK) overexpression and reversal of nilotinib resistance through inhibition of phosphor-JNK (p-JNK) by metformin in nilotinib-resistant cells. (A) Immunoblotting analysis of nilotinib-sensitive and nilotinib-resistant cells for determining endogenous MAP kinase expression (ERK, JNK, and p38) (B) Cell proliferation in response to nilotinib alone and in combination with a JNK inhibitor in nilotinib-resistant cell lines. EZ-Cytox assays were performed using triplicate samples. (C) Western blotting analysis of cells after treatment with indicated concentrations of metformin for 48 hours. (D) Cell proliferation in response to nilotinib alone and in combination with metformin in nilotinib-resistant cell lines. ap < 0.05, bp < 0.01, cp < 0.001, respectively.
Combined treatment with nilotinib and metformin decreases phosphor-JNK expression, overcomes nilotinib resistance, and induces apoptosis through caspase activation in nilotinib-resistant CML cells.
After treating the cells with 10 mM metformin and 10 nM nilotinib for 48 hours, we measured apoptosis induction by measuring the levels of PARP, caspase 3, caspase 9, and Bcl-xL, which are hallmark proteins of apoptosis. Neither metformin nor nilotinib alone induced PARP, caspase 3, or caspase 9 cleavage in nilotinib-resistant cells. However, the combination therapy induced PARP, caspase 3, and caspase 9 cleavage. Furthermore, the combination treatment notably decreased the expression of phosphorylated JNK in nilotinib-resistant cells compared to control. These data indicated that JNK downregulation by metformin reversed nilotinib resistance (Fig. 4A). Similar results were observed by trypan blue exclusion assay (Fig. 4B). The early apoptotic cell percentage increased by seven-or eight-fold compared to control (Fig. 4C). Finally, we confirmed whether the apoptosis-inducing effect of the combination treatment was dependent on caspase. Combined treatment with metformin and nilotinib markedly enhanced apoptosis signaling, whereas combined treatment with metformin, nilotinib, and z-VAD-fmk (25 μM), a pan-caspase inhibitor, significantly attenuated apoptosis signaling (PARP, caspase 3 and caspase 9 cleavage). These data implied that the apoptosis-enhancing effect of combination treatment with metformin and nilotinib was mediated via increased caspase activation (Fig. 4D).

Combined treatment with nilotinib and metformin induced apoptosis by decreasing Bcl-xL expression in nilotinib-resistant chronic myeloid leukemia cells. (A) Proteins from nilotinib-sensitive and nilotinib-resistant cells were solubilized and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. (B) Cell death in nilotinib-resistant cell lines was analyzed by trypan blue exclusion assay after drug treatment. (C) Cells were stained with annexin V and propidium iodide, followed by flow cytometry. (D) Cells were pretreated with 25 µM z-VAD-fmk for 30 minutes, and then incubated with 10 mM metformin and 10 nM nilotinib for 48 hours. Immunoblotting analysis of nilotinib-resistant cell lines for c-Jun N-terminal kinase (JNK), phosphor-JNK (p-JNK), PARP, caspase 3, and caspase 9 was performed with actin as a loading control. ap < 0.05, bp < 0.01, cp < 0.001, respectively.
DISCUSSION
Nilotinib is a second-generation TKI designed from its parent compound imatinib, and 30 times more potent than imatinib for CML [18,19]. It is also active against ABL, KIT, PDGFR-a, PDGFR-b, and Src family kinases. We developed nilotinib-resistant cell lines by culturing the cells with gradually increasing concentrations of nilotinib for over 6 months. A similar approach was applied by other groups to develop resistance in leukemia cells [20,21]. Although we generated nilotinib-resistant cell lines with low dose (10 nM) nilotinib, the IC50 of the nilotinib-resistant cells was six times higher than that of the parental cells (Fig. 2A). We also showed differences in nilotinib-sensitive and nilotinib-resistant cells by DAPI nuclear staining, trypan blue assay, and western blotting (Fig. 2B-2D).
Metformin is a biguanide widely used for treating type 2 diabetes. Recent studies indicated that metformin plays an anticancer role in several cancer types. For instance, metformin exerts anticancer effects through inhibition of the Shh signaling pathway in breast cancer and the HIF1 a pathway in gastric cancer [8,10]. Nevertheless, the effect of metformin on CML has not been elucidated. Because nilotinib is well known to cause an adverse effect of hyperglycemia, investigation of the effect of metformin combined with nilotinib would be clinically relevant. Our results suggested that metformin alone was not significantly effective, but combined treatment with metformin and nilotinib synergistically induced apoptosis in CML cell lines (Fig. 1).
The JNK family is a subfamily of mitogen-activated protein kinases (MAPK). JNK has also been correlated with pathological conditions, such as inflammation, neurodegenerative diseases, and cancers [22-24]. Interestingly, oncology studies have shown that JNK has a dual function to allow both cell survival and cell death. Activating the proto-oncogene c-Jun shows the function of JNK in oncogenic signaling, and in contrast, JNK induces the defense mechanisms to protect organisms against various situations by regulating stress-induced apoptosis and autophagy as a tumor suppression signaling [25]. Deregulation or activation of this signal is often observed in human cancers; but to our knowledge, this was the first time that it was shown as a nilotinib resistance mechanism.
In this study, we identified the mechanism responsible for nilotinib resistance in two CML cell lines (K562 and KU812). Although there have been several studies reporting the association of Syn, Lyn, and Src proteins with nilotinib resistance [21,26], this was the first study to investigate the overexpression of JNK in nilotinib-resistant cell lines. Our study indicated that nilotinib resistance involved the upregulation of JNK expression, and we confirmed this by restoring the sensitivity to nilotinib using a JNK inhibitor. Targeting JNK signaling by a JNK inhibitor, SP600125, transformed nilotinib-resistant cells into nilotinib-sensitive cells, suggesting that JNK promoted cell survival signaling to maintain resistance to nilotinib. We also checked the expression of other MAP kinase signaling proteins, such as ERK and p38 MAPK, but there were no observable changes (Fig. 3A).
Metformin, which has anticancer effects, also regulates JNK signaling. For example, metformin inhibits the activities of ERK1/2 and JNK1/2 in hepatocellular carcinoma [27]. We have checked whether metformin regulates JNK overexpression in nilotinib-resistant CML cells, and we have found that it decreased JNK phosphorylation not only in nilotinib-sensitive cells but also in nilotinib-resistant cells (Fig. 3C). However, owing to the up-regulated JNK expression in nilotinib-resistant cells, JNK expression in nilotinib-resistant cells was significantly decreased by metformin compared to that in nilotinib-sensitive cells. Finally, nilotinib could act as a TKI after JNK expression was decreased by metformin in nilotinib-resistant cell lines. Combination treatment with nilotinib and metformin had greater efficacy than treatment with nilotinib and a JNK inhibitor, SP600125. We also discovered that metformin increased the efficacy of nilotinib through reduction of Bcl-xL (Fig. 1D).
In conclusion, metformin was effective in decreasing phosphorylated JNK levels, resulting in the restoration of nilotinib sensitivity. Combined treatment with nilotinib and metformin was more effective than combined treatment with nilotinib and JNK inhibitor in terms of cell proliferation inhibition. This study suggested that the combination of metformin and nilotinib may have clinical benefits of enhancing antileukemia efficacy and overcoming resistance to nilotinib.
KEY MESSAGE
1. Metformin enhances nilotinib-mediated apoptosis in chronic myeloid leukemia (CML) cells.
2. Up-regulated phosphor-c-Jun N-terminal kinase (JNK) is a phenotype of acquired resistance to nilotinib in CML cells.
3. Metformin decreases up-regulated phosphor-JNK and restores anticancer effects of nilotinib in nilotinib-resistant CML cells.
4. The combination therapy with metformin and nilotinib is suggested as a useful strategy to treat nilotinib-resistant CML patients with high blood glucose level.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by the Brain Korea (BK) 21 Plus Program.