Consensus regarding diagnosis and management of atypical hemolytic uremic syndrome
Article information

Abstract
Thrombotic microangiopathy (TMA) is defined by specific clinical characteristics, including microangiopathic hemolytic anemia, thrombocytopenia, and pathologic evidence of endothelial cell damage, as well as the resulting ischemic end-organ injuries. A variety of clinical scenarios have features of TMA, including infection, pregnancy, malignancy, autoimmune disease, and medications. These overlapping manifestations hamper differential diagnosis of the underlying pathogenesis, despite recent advances in understanding the mechanisms of several types of TMA syndrome. Atypical hemolytic uremic syndrome (aHUS) is caused by a genetic or acquired defect in regulation of the alternative complement pathway. It is important to consider the possibility of aHUS in all patients who exhibit TMA with triggering conditions because of the incomplete genetic penetrance of aHUS. Therapeutic strategies for aHUS are based on functional restoration of the complement system. Eculizumab, a monoclonal antibody against the terminal complement component 5 inhibitor, yields good outcomes that include prevention of organ damage and premature death. However, there remain unresolved challenges in terms of treatment duration, cost, and infectious complications. A consensus regarding diagnosis and management of TMA syndrome would enhance understanding of the disease and enable treatment decision-making.
INTRODUCTION
Thrombotic microangiopathy (TMA) describes the histologic abnormalities in the endothelium found in a number of conditions. In 1924, Moschowitz reported that thrombotic thrombocytopenic purpura (TTP) could be distinguished by hyaline thrombi in multiple organs, including the kidneys. In 1952, Symmers recognized pathognomonic disseminated lesions that occurred only in microscopic vessels and described the condition as TMA [1]. Subsequently, Gasser et al. [2] described hemolytic uremic syndrome (HUS) in 1955. However, the pathogeneses of TTP, HUS, and TMA remained unclear until the 1980s. The pathogenesis of TTP—i.e., atypically large von Willebrand factor multimers—was discovered only in 1982 [3]. In 1985, Shiga toxin produced by Escherichia coli was found to cause HUS [4]. The pathogenesis of TMA was recently established as endothelial cell injury associated with alterations in factors that affect angiogenesis, coagulation, platelet activation, and complement function [1].
TMA syndromes are defined by specific clinical characteristics, including microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and pathologic evidence of endothelial cell damage; these manifestations lead to ischemic end-organ injuries [1]. Shiga toxin-associated HUS (i.e., typical HUS) is a TMA syndrome caused by infection with Shiga toxin-producing E. coli (STEC) or Shigella. In contrast, atypical HUS (aHUS) is caused by a genetic or acquired defect in regulation of the alternative complement pathway [5]. Manifestations of atypical HUS can range from mild hematologic abnormalities to severe (sometimes life-threatening) end-organ damage such as gastrointestinal bleeding, seizure, blindness, or acute kidney injury requiring dialysis. Secondary TMA syndromes may be characterized by clinical manifestations similar to those of aHUS; however, these TMA syndromes typically have specific causes.
Differentiating among TMA syndromes is difficult but crucial because of the need for distinct therapeutic approaches. Previously, distinguishing between TTP and HUS was problematic; however, this was resolved by measurement of the serum levels or activities of von Willebrand factor-cleaving protein, a metalloproteinase with thrombospondin type 1 motif, member 13 (ADAMTS13). Currently, distinguishing between aHUS and secondary TMA syndromes remains challenging. The two conditions have similar manifestations but different treatment strategies. Secondary TMA syndromes can be managed by removal of triggering factors, whereas aHUS requires correction of complement dysregulation and removal of triggering factors. Moreover, eculizumab—a recombinant, humanized, monoclonal immunoglobulin that targets complement component 5 (C5) and hinders the cleavage of C5 to C5a and C5b—was approved for the treatment of aHUS, but not secondary TMA syndromes, as an alternative to plasmapheresis [6-8].
This is a consensus report of South Korean experts, including hematologists, adult and pediatric nephrologists, transplantation surgeons, pathologists, and genetic laboratory medicine specialists. This expert group gathered for several meetings from March 2019 to November 2019 to share and harmonize opinions regarding diagnosis and management of TMA syndromes. During these meetings, we generated updated consensus TMA diagnostic criteria and triggering factors based on the 2016 South Korean clinical practice guidelines for aHUS [9]. Here, we suggest a definition and differential diagnosis for TMA syndromes based on clinical manifestations and genetic factors. We also summarize therapeutic strategies for aHUS in both adult and pediatric patients.
TMA DIAGNOSTIC CRITERIA
Hematologic abnormalities
TMA is defined on the basis of clinical, laboratory, and pathological features. The laboratory features include MAHA characterized by fragmented erythrocytes (e.g., schistocytes or helmet cells), thrombocytopenia (with increased numbers of bone marrow megakaryocytes), an elevated serum level of lactate dehydrogenase (LDH), and abnormal laboratory results related to TMA-mediated organ damage (e.g., increased creatinine level and hematuria or proteinuria). The pathological features are systemic microvascular thrombosis and endothelial injury (Fig. 1) [1,10].

Differential diagnosis of thrombotic microangiopathy syndromes. LDH, lactate dehydrogenase; RBC, red blood cell; AKI, acute kidney injury; GI, gastrointestinal; CNS, central nervous system; CV, cardiovascular; STEC HUS, Shiga-toxin producing Escherichia coli hemolytic uremic syndrome; ADAMTS13, metalloproteinase with thrombospondin type 1 motif, member 13; TTP, thrombotic thrombocytopenic purpura; SCr, serum creatinine; BMT, bone marrow transplantation.
According to the definition of TMA suggested by the Korean aHUS Working Group, evidence of MAHA (hemoglobin level < 10 g/dL, increased serum LDH level, decreased serum haptoglobin level, and presence of red blood cell fragments in a peripheral blood smear) and thrombocytopenia (platelet count < 150,000/μL) are required for the diagnosis of TMA [9]. The Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society suggested similar criteria for the diagnosis of TMA: MAHA (confirmed based on an increased serum LDH level, a marked reduction in serum haptoglobin level, and the presence of red blood cell fragments) with a hemoglobin level of < 10 g/dL and thrombocytopenia (platelet count < 150,000/μL) [11,12]. However, a haptoglobin level below the lower limit of normal (LLN) or the presence of schistocytes may not be observed, despite the presence of active TMA. In addition, the platelet count may be within the normal range in up to 20% of patients with aHUS [13] and a hemoglobin level above the LLN may be observed in patients with TMA [13,14]. From these, we suggest the previous Korean definition for TMA is changed. The laboratory criteria for TMA applied in previous clinical trials for aHUS were: (1) evidence of hemolysis such as an LDH level above the upper limit of normal, a haptoglobin level below the LLN, or the presence of schistocytes on a peripheral blood smear; and (2) low platelet count (< 150,000/μL) or a 25% reduction in the average of three platelet counts before the most recent TMA complication [15].
Therefore, the laboratory criteria for TMA should include two categories such as evidence of MAHA with a serum hemoglobin level below the LLN and thrombocytopenia (below the LLN or a reduction of > 25% from the patient’s usual baseline). The evidence of MAHA includes an increased serum LDH level and the presence of red blood cell fragments. However, the schistocyte criterion for MAHA may be ignored in patients with definite clinical or pathologic evidence of TMA.
Kidney manifestations
Any organ that contains endothelial cells can be affected by TMA. Glomerular endothelial cells are one of the main targets of TMA for a reason that remains unclear. The fenestrated glomerular endothelium may lack complement regulators, increasing its susceptibility to complement activation [16]. Alternatively, podocyte injury may lead to endothelial injury, because the health of glomerular endothelial cells is dependent on podocyte- derived vascular endothelial growth factor [17].
Pathologic findings that reflect tissue responses to endothelial injury in the kidney are classified as follows, according to activity, microvascular area involved, and mechanism: active versus chronic, glomeruli versus arterioles versus arteries, and thrombotic versus non-thrombotic lesions. These criteria were developed during the Kidney Disease Improving Global Outcomes Controversies Conference for aHUS and complement component 3 (C3) glomerulopathy [18]. Active lesions include intravascular fibrin thrombi with mucoid changes, as well as endothelial swelling. In addition, swollen glomerular endothelial cells with loss of fenestration, expansion of lamina rara interna, and fibrin tactoid with platelets and fragmented red blood cells can be found on electron microscopic examination [19]. Chronic lesions contain double contours of capillary walls, mesangial interposition in glomeruli, hyaline deposits in arterioles, and fibrous intimal thickening with concentric lamination (onion skin appearance) in arteries. Thrombotic lesions feature intraluminal fibrin or fibrin-platelet plugging. Although it is difficult to identify the etiology of TMA based on kidney pathology findings, recognition of the TMA pattern of kidney injury is crucial to provide firm evidence of TMA and to identify a potential underlying mechanism.
The above pathologic injuries typically manifest as acute kidney injury or urinary abnormalities. Acute kidney injury is defined as a rapid decline in kidney function characterized by a serum creatinine elevation by ≥ 0.3 mg/dL within 48 hours or ≥ 1.5-fold the baseline value, which is known or presumed to have occurred within the prior 7 days [20]. In urinalysis, albuminuria is identified in more than half of patients with TMA [21]. It is unknown whether the amount of proteinuria or microalbuminuria correlates with the severity of TMA syndrome or not.
Involvement of other organs
Gastrointestinal tract involvement is frequently found in TMA. Diarrhea, which may be bloody, is the most common feature of gastrointestinal involvement. Furthermore, the central nervous system is able to be affected in TMA; this is characterized by confusion, seizure, stroke, or encephalopathy. These manifestations were previously regarded as unique characteristics of TTP. Respiratory tract involvement is a common presentation of TMA syndrome; this includes upper respiratory tract infection, pulmonary hypertension, and pulmonary hemorrhage. The cardiovascular system has abundant endothelial cells. Therefore, hypertension including malignant hypertension frequently occurs in TMA patients. In addition, large vessels (e.g., carotid, cerebral, coronary, and pulmonary) and myocardial vessels themselves may be damaged by TMA syndromes. Skin damage has been reported as purpuric or necrotic changes and ocular involvement, including severe retinal detachment due to retinal artery occlusion, although, such damage is rare. These extrarenal TMA manifestations are rarely demonstrated pathologically because of the difficulty in involved tissue acquisition. Therefore, understanding the kidney pathology and associated molecular mechanisms may enable differentiation among TMA syndromes with various causes.
Diagnosis of pediatric aHUS
When a child presents with HUS, the first condition to consider is diarrhea-associated HUS. Diarrhea-associated HUS is typically a result of enterohemorrhagic STEC, and the patient often exhibits prodromal symptoms of diarrhea, abdominal pain, and hematochezia. STEC-HUS is diagnosed when STEC is cultured from stool or Shiga toxin is identified by polymerase chain reaction. HUS in a previously healthy child, particularly an infant, without specific prodromal symptoms of gastrointestinal infection is presumably aHUS.
While diagnostic criteria for TMA and HUS in children are identical those in adults, the conditions to exclude before diagnosis of aHUS are relatively different. In adults, TTP is one of the first differential conditions after secondary TMA syndromes; therefore, ADAMTS13 activity should be measured first, along with investigations of medications and conditions that can induce TMA such as malignancy and autoimmune diseases. Secondary TMA syndromes should also be considered in children; however, the majority of pediatric patients present without a medical history suggestive of secondary TMA syndromes, and TTP is uncommon. Therefore, previously healthy children with HUS who exhibit no evidence of gastrointestinal infection should be assumed to have aHUS. After assessment of ADAMTS13 activity and measurement of complement factors and their antibodies, treatment targeting aHUS should be administered rapidly.
In addition, aHUS typically occurs in the presence of a trigger that activates the complement system. Notably, infection or another factor that causes activation of the immune system can trigger aHUS. A considerable number of patients with aHUS, particularly those with anti-complement factor H (CFH) antibodies, have gastrointestinal symptoms. Therefore, the presence of gastrointestinal symptoms cannot be used to rule out aHUS. If the course is atypical for diarrhea-associated HUS and gastrointestinal symptoms are mild while HUS is severe, aHUS should be suspected.
Key message
• Evidence of MAHA (increased serum LDH level and presence of red blood cell fragments) with a serum hemoglobin level below the LLN and thrombocytopenia (below the LLN or a reduction of > 25% from the patient’s usual baseline) should be included for the diagnosis of TMA. However, the schistocyte criterion for MAHA may be ignored in patients with definite clinical or pathologic evidence of TMA.
• Atypical HUS can range from mild hematologic abnormalities to severe (sometimes life-threatening) end-organ damage such as gastrointestinal bleeding, seizure, blindness, or acute kidney injury requiring dialysis.
• In patients with TMA and any organ injury, assays for STEC and ADAMTS13 activity should be performed for differential diagnosis. aHUS should be suspected in all STEC-negative patients with normal ADAMTS13 activity (≥ 10%).
• Pediatric patients who exhibit TMA without gastrointestinal infection should be assumed to have aHUS and should be treated rapidly because TTP is uncommon in children.
TRIGGERS OF TMA
Because of the incomplete genetic penetrance of aHUS, a second hit, which may be genetic or acquired, is required for its development. A variety of medications and clinical conditions such as infection, pregnancy, malignancy, and autoimmune disease can be associated with TMA syndromes. These triggering conditions are considered causes of secondary TMA syndromes. However, recent evidence suggests that they cannot be separated from aHUS. Rather, they may coexist with aHUS, and may contribute to triggering activation of the complement system in patients genetically predisposed to aHUS [22]. For that reason, they are regarded as co-existing diseases or as complement-amplifying conditions. TMA-associated conditions, co-existing diseases, and complement-amplifying conditions are described as triggering factors in this article. If TMA persists despite remission or successful treatment of triggering factors, then the diagnosis of aHUS should be strongly considered [22]. According to the report by Noris et al. [23] regarding the clinical characteristics of 273 patients with aHUS, 70% had at least one triggering factor; this was most commonly diarrhea/gastroenteritis (16.5%), followed by upper respiratory tract infection (12.8%).
Pregnancy
Most cases of pregnancy-associated TMA develop during the postpartum period [24]. In one study, the renal outcomes were very poor, such that 76% of the patients progressed to end-stage renal disease (ESRD) despite plasmapheresis treatment. Recently, complement mutations were identified in more than 50% of patients with pregnancy-associated TMA. Therefore, pregnancy can be regarded as a triggering factor for TMA syndromes in women with underlying genetic abnormalities associated with complement system regulation [24]. Furthermore, these findings suggest that eculizumab may be an effective therapy for pregnancy-associated TMA syndromes in women with dysregulated complement activation [25].
Malignant hypertension
Although the endothelial damage caused by TMA syndrome can induce elevation of blood pressure, severe hypertension may lead to further endothelial damage and changes in the renal vasculature, including fibrinoid necrosis of arterioles and glomerular capillary tufts [26]. Therefore, patients with hypertension-associated TMA should undergo aggressive blood pressure control and receive supportive treatment to resolve acute features of TMA and prevent further kidney injury [27]. In a recent retrospective case series, genetic predisposition to preferentially use the alternative complement pathway was identified in patients for whom TMA was initially triggered by severe hypertension; among nine patients, eight demonstrated progression to ESRD despite controlled blood pressure [26]. This suggests that dysregulation of the alternative complement pathway is the cause of renal impairment in patients with malignant hypertension.
Kidney transplantation
The incidence of post-transplant TMA is reportedly 5.6 cases per 1,000 kidney transplant (KT) recipients [28]. Post-transplant TMA can occur at any time, but most commonly manifests at 3 to 6 months after KT [29,30]. The incidence of post-transplant TMA was 36.5-fold greater than that of TMA in patients with native kidney [28]. The risk factors for TMA in KT recipients are more complex than those in patients with other conditions (Fig. 2) [31-34].

Treatment approach for post-transplant thrombotic microangiopathy. Modified from Campistol et al. [36]. TMA, thrombotic microangiopathy; ADAMTS13, metalloproteinase with thrombospondin type 1 motif, member 13; STEC, Shiga toxin-producing Escherichia coli; DIC, disseminated intravascular coagulation; AMR, antibody mediated rejection; CMV, cytomegalovirus; PE, plasma exchange.
Genetic mutations are also important risk factors for the recurrence of aHUS during the post-transplant period. Approximately 50% of KT recipients with genetic mutations developed ESRD at 3 years after KT, due to recurrent TMA [35]. Therefore, living related donor KT is not recommended due to the possibility of hidden genetic mutations.
The majority of patients with recurrent TMA may exhibit recurrence of aHUS [31]. Recurrent TMA has more sudden onset, more severe clinical features, a higher rate of systemic involvement, and worse graft outcomes than de novo TMA [36]. In KT recipients, recurrent TMA with possible recurrence of aHUS should be suspected in the presence of the following: (1) hypertensive nephrosclerosis in kidney biopsy before KT or ESRD of unknown etiology; (2) young age; (3) occurrence soon after KT; (4) unexplained severe and rapid progression of clinical features after KT; and (5) clinical manifestations without evidence on kidney biopsy [32,36,37]. However, some patients with post-transplant TMA do not show systemic signs; for such patients, allograft biopsy is needed to confirm the diagnosis [37].
Other solid organ and bone marrow transplantation
TMA can also develop after transplantation of other solid organs, including the liver, pancreas, lung, and heart [38,39]. The underlying mechanism has not been elucidated; it is likely to be multifactorial, involving calcineurin inhibitors, viral infections, and antibody-mediated rejection. Allogeneic bone marrow transplants can also cause secondary TMA syndromes. Similarly, the causes of TMA after bone marrow transplant are likely to be multifactorial; these may include graft versus host disease, human leukocyte antigen mismatch, chemotherapy, radiation therapy, infection, and the use of calcineurin inhibitors [40-42]. Meticulous supportive care and removal of the precipitating factors are the best treatment options for TMA in such patients.
Drugs
Development of TMA has been reported in relation to various drugs, including quinine, cyclosporine, tacrolimus, sirolimus, bevacizumab, gemcitabine, interferon, mitomycin C, oxaliplatin, and sunitinib [43-49]. Drug-induced TMA develops by two main mechanisms: immune-mediated and direct toxicity-mediated. In immune-mediated drug-induced TMA, the drug induces antibodies that react with platelets and endothelial cells in a distinctive manner. The most common cause of immune-mediated drug-induced TMA is quinine [50]. Toxicity-mediated TMA may have multiple mechanisms, and is often dose-dependent. Common causes of toxicity-mediated drug-induced TMA include type 1 interferon [51], calcineurin inhibitors, and a variety of anticancer drugs [52] including bevacizumab, sunitinib, and gemcitabine. Thus far, there are no definite guidelines for the treatment of drug-induced TMA; supportive care and discontinuation of the offending drug are the primary treatment approaches used [25]. A better understanding of the mechanisms of drug-induced TMA is needed to enhance diagnosis and treatment and prevent re-exposure to the offending drug.
Autoimmune disorders and vasculitides
Systemic lupus erythematosus-associated TMA is related to disease activity. Immune complex formation and sequential terminal complement activation constitute an important pathogenetic mechanism of organ involvement, including the kidney. Concomitant TMA in patients with lupus nephritis is associated with higher susceptibility to infection, worse kidney outcomes, and a lower survival rate [22]. In one study, use of eculizumab resulted in improvement of symptoms, renal function, and cytopenia in patients who were refractory to conventional treatment for systemic lupus erythematosus [53]. Advanced scleroderma can also be associated with aHUS, particularly in cases of scleroderma renal crisis. There is also an anecdotal report that eculizumab treatment led to resolution of TMA in a patient with scleroderma renal crisis [54].
Associations of TMA and systemic autoimmune vasculitis—such as eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), granulomatosis with polyangiitis (Wegener granulomatosis), and Henoch-Schoenlein purpura—have been reported [55]. Some TMA syndromes associated with autoimmune conditions are essentially aHUS triggered by autoimmunity, and therefore benefit from complement inhibition [56].
Fibrinous thrombotic diseases
Disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, and catastrophic antiphospholipid syndrome are differential diagnoses of aHUS. However, in some instances, those fibrinous thrombotic conditions can trigger aHUS due to crosstalk between the complement and coagulation systems: the complement cascade is activated during activation of coagulation cascades [57]. Disseminated intravascular coagulation-associated TMAs, often observed in bone marrow infiltration by solid tumors or in liver failure, differ from aHUS. However, there may be some overlap [58] based on crosstalk between the two systems.
Infection and sepsis
TMA can be triggered by viral infections, including influenza virus, human immunodeficiency virus, adenovirus, and cytomegalovirus (often in the post-transplant setting) [59]. In addition to toxin-mediated TMA due to STEC, systemic bacterial infection can trigger complement-mediated TMA. In patients with Streptococcus pneumoniae infection, bacterial neuraminidase removes neuraminic acid from red blood cells, platelets, and glomerular endothelial membranes, thereby exposing the Thomsen-Friedenreich antigen (T-antigen; CD176) to natural antibodies. The antigen-antibody complexes thus formed activate the classical complement pathway [14]. A similar mechanism is proposed for Pseudomonas aeruginosa-related TMA [60]. Favorable outcomes of eculizumab have been reported in TMA associated with the aforementioned infections, including cytomegalovirus [61].
Sepsis can trigger TMA because it is a strong complement-activating condition [62]. In patients with sepsis, excessive production of C5a results in paralysis of innate immunity and contributes to failed infection responses [62]. A potential benefit of eculizumab for improving sepsis outcomes has been suggested in a case report [63].
Key message
• A variety of medications and clinical conditions (e.g., infection, pregnancy, malignancy, and autoimmune disease) are associated with TMA syndromes.
• TMA syndromes may coexist with aHUS and may contribute to complement system activation in patients genetically predisposed to aHUS.
GENETIC TESTING IN aHUS
Genetic variants leading to inappropriate activation of the alternative complement pathway by loss or gain of protein function cause predisposition to aHUS, with or without triggering factors [64]. Overall, genetic aHUS (i.e., aHUS with identifiable predisposing variants in disease-associated genes) constitutes approximately 60% of all cases of aHUS [16,65,66]. The genes associated with aHUS include CFH, CD46 or membrane cofactor protein (MCP), C3, complement factor I (CFI), complement factor B (CFB), thrombomodulin (THBD), plasminogen (PLG), and diacylglycerol kinase epsilon (DGKE). Genetic testing for aHUS by massively parallel sequencing has recently become available as a clinical test in South Korea, and is indicated for all patients suspected to have aHUS without evidence of STEC infection, severe ADAMTS13 deficiency, or hyperhomocysteinemia/methyl-malonic aciduria. Genetic information supports diagnosis, clarity regarding the underlying pathophysiology, and guides treatment decisions; moreover, it is predictive of prognosis [67]. However, the absence of variants of a given set of genes does not preclude the diagnosis of aHUS because of the possibility of (1) variants in genes other than classical aHUS genes and (2) variants that can be missed by high-throughput sequencing (e.g., large genomic rearrangements or intronic variants not covered by conventional panel testing). Comprehensive analysis involving an extended set of genes can uncover other genes/variants associated with aHUS. In addition, most variants (except those in DGKE) that cause autosomal recessive aHUS in pediatric patients have incomplete penetrance. The disease can be triggered by environmental conditions in genetically susceptible patients [23]. Therefore, the diagnosis of aHUS depends primarily on clinical findings along with laboratory tests for differential diagnosis, such as tests for STEC and ADAMTS13 activity. Lastly, interpretation of variants of uncertain significance, as described in the American College of Medical Genetics and Genomics guidelines, should be done cautiously in aHUS, considering the possible contributions of variants that exist in the population and those predicted to be benign or tolerated by in silico analyses without laboratory evidence of functional consequences [68,69].
Copy number analysis by multiplex ligation-dependent probe amplification is needed to detect homozygous deletions involving complement factor H related protein 1 (CFHR1)/complement factor H related protein 3 (CFHR3) genes, which are associated with auto-antibodies to CFH, particularly in pediatric patients [70,71]. Multiplex ligation-dependent probe amplification can also detect CFH/CFHR1 or CFHR1/CFH hybrids generated by genomic rearrangement [72]. Lastly, targeted genotyping is indicated for the family members of aHUS patients with genetic variants to assess the associations of variant(s) of interest with the disease, to guide treatment decisions, and to determine their genetic profiles. Careful genetic counseling is warranted due to the incomplete penetrance of the disease, especially when multiple variants are present in the family. In summary, genetic analysis in the form of a multi-gene panel test is essential for all patients suspected to have aHUS, along with multiplex ligation-dependent probe amplification analysis and an anti-CFH autoantibody test, particularly in pediatric patients. The interpretation of genetic test results should be cautious and should consider the disease characteristics.
In South Korea, a multicenter cohort of 51 Korean children with aHUS was screened for genetic variants using targeted exome sequencing covering 46 complement-related genes, together with measurement of the anti-CFH autoantibody titer [73]. Disease-associated variants were detected in 11 patients (22%); the affected genes were CFH, CFI, CD46 (MCP), and DGKE. Twelve patients (24%) had a homozygous deletion in the CFHR1 gene. The prevalence of anti-CFH autoantibody was 29%, higher than that in other regions, and 73% of patients with anti-CFH autoantibody had a homozygous deletion in the CFHR1 gene. Therefore, all Korean children with suspected aHUS should be tested for the presence of anti-CFH antibodies in blood samples collected before treatment.
A multicenter study involving 66 adult South Korean patients with aHUS demonstrated a variant detection rate of approximately 60% by exome sequencing and screening of 14 genes [74]. The distribution and frequency of genetic variants and homozygous CFHR1 deletion revealed a markedly different genetic profile, compared with that of South Korean pediatric patients with aHUS, as well as that of patients of other ethnicities. More diverse genes were affected than in adult South Korean patients than in pediatric South Korean patients, and homozygous deletion of CFHR1 was rare. The higher frequency of THBD variants was a remarkable finding and was regarded as an ethnic difference.
Key message
• Genes associated with aHUS include CFH, CD46 or MCP, C3, CFI, CFB, THBD, PLG, and DGKE.
• Genetic information supports diagnosis, clarity regarding the underlying pathophysiology, and guides treatment decisions; moreover, it is predictive of prognosis. However, the absence of variants of a given set of genes does not preclude the diagnosis of aHUS.
THERAPEUTIC STRATEGIES FOR aHUS
Therapeutic strategies for aHUS have been based on resolution of dysregulation in the complement system. Previously, plasma therapy, including plasma infusion and exchange, was the first-line treatment option for patients with aHUS. However, the therapeutic efficiency of plasma therapy varies according to the causative genetic abnormalities of the alternative complement pathway or other pathways. Liver transplantation has been considered an alternative treatment option, although its utility is limited due to perioperative morbidities and donor shortage. Eculizumab, a monoclonal antibody to terminal C5, was introduced for the management of this debilitating disease and has shown superior outcomes; notably, it prevents organ damage and premature death more effectively than prior modalities. Eculizumab has contributed to control of aHUS and has facilitated clinical, functional, and molecular research to accelerate related diagnosis and treatment. The long-acting C5 complement inhibitor, ravulizumab, received early approval by the Food and Drug Administration for the treatment of aHUS. This advance will improve the quality of life of aHUS patients.
Plasma therapy
Empiric plasma therapy in aHUS remains recommended, pending Shiga toxin, ADAMTS13, anti-CFH autoantibody, and genetic testing [14,36,75]. Plasma therapy can be administered as a plasma infusion or plasma exchange (PE). Functional complement-regulating proteins can be supplied by plasma infusion with fresh frozen plasma. The effect of plasma infusion is limited in a small number of patients with complete CFH deficiency [36]; in contrast, PE can replace absent or defective complement regulators and can remove autoantibodies or mutated circulating complement regulators [76]. Before a firm diagnosis is made, PE is regarded as the standard of care. When TTP, STEC HUS, and secondary TMA syndromes have been ruled out, a switch to eculizumab is recommended [18]. When eculizumab is unavailable, therapeutic PE remains an alternative treatment option.
The American Society for Apheresis adjusted the category for therapeutic apheresis from II (second-line therapy) to III (non-established role of apheresis) in patients with complement factor gene mutations [75,77]. For CFH autoantibody-related aHUS, the combination of PE and immunosuppression has been effective (category I; firstline therapy). PE is not considered effective for patients with MCP mutations, as MCP is a non-circulating protein attached to the cell membrane (category III) [75].
Evaluation of the response to plasma therapy
If eculizumab is not applicable, a 1.5 plasma volume, equivalent to 60 to 75 mL/kg per session, should be exchanged for fresh frozen plasma. PE must be performed daily for 5 days, then five times weekly for 2 weeks, and subsequently three times weekly for 2 weeks [78]. Longterm maintenance therapy can be weekly to every 2 to 4 weeks [79]. The absence of a PE response can be defined as either the absence of normalization of platelet count or the absence of reduction in plasma creatinine level ≥ 25% in patients without severe chronic damage on biopsy or renal imaging after 5 consecutive days of PE [14,80]. However, the number of sessions and duration of PE for aHUS are controversial. Decisions concerning duration or discontinuation should be made based upon the patient’s response and condition [75].
Interpretation of kidney dysfunction after hematologic normalization
It is uncertain whether kidney dysfunction can be considered a marker of disease activity. In an observational study of complement abnormalities in 273 patients with aHUS, 53% to 67% of the patients developed ESRD during the first episode or within 3 years of onset, although plasma therapy induced complete or partial remission of 63%, 25%, 57%, 88%, and 75% of episodes in patients with CFH, CFI, C3, or THBD mutations or anti-CFH autoantibodies, respectively [23]. The majority of patients with CFH, CFI, C3, or THBD mutations or anti-CFH autoantibodies lost renal function or died during the presenting episode, or demonstrated progression to ESRD as a result of relapses after PE [23,79,81]. However, it remains unclear whether persistent or progressive kidney function deterioration despite plasma therapy reflects unresolved active kidney inflammation or merely chronic ischemic changes.
Eculizumab
Eculizumab, a monoclonal antibody that blocks the cleavage of C5, is the first-line treatment for patients with aHUS [82]. Early initiation of eculizumab has been effective for minimizing renal damage [83]. If eculizumab is unavailable, PE and/or plasma infusion can be considered.
The dosage and schedule of eculizumab should be adjusted according to body weight, as in Table 1 [9]. Patients should receive meningococcal vaccination at least 2 weeks prior to the first dose of eculizumab because the drug increases the risk of meningococcal infection. If eculizumab treatment is initiated less than 2 weeks after vaccination, patients should receive additional prophylactic antibiotics until 2 weeks after vaccination [84].
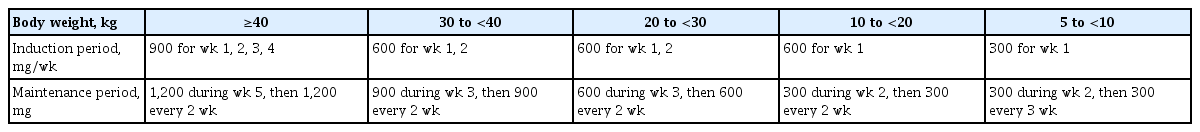
Recommended dosage and schedule of eculizumab for patients with atypical hemolytic uremic syndrome
Ravulizumab, a long-acting C5 inhibitor, was approved by the Food and Drug Administration for the treatment of adult and pediatric patients ≥ 1 month of age with aHUS to inhibit complement-mediated TMA. Ravulizumab treatment of aHUS should be maintained for a minimum duration of 6 months; extension beyond 6 months should be determined on an individual basis. Ravulizumab reduces the treatment burden, with infusions limited to once every 4 or 8 weeks depending on body weight. Further studies are needed regarding its use in pregnant women [85].
Kidney transplantation
Prevention of TMA after KT
After KT, the recurrence rate of aHUS is high (approximately 60%) and that of graft survival is poor (approximately 90%) [86]. Advances in genetics and the development of eculizumab have altered the preparation and treatment of patients with aHUS after KT.
Candidates for KT who have genetic mutations associated with aHUS require some precautions in accordance with risk stratification based on the mutated gene. Patients with MCP, DGKE, undetectable anti-CFH autoantibody, and no mutation are regarded as lowrisk for post-transplant TMA. In contrast, patients with CFH, CFI, C3, CFB, CFH/CFHR1 hybrid, or combined mutations and no mutation are regarded as moderateto-high-risk for aHUS recurrence [18,31,34,36]. For the latter group of patients, the following donors should be excluded [34,36]: (1) positive cross-match or donor-specific antibody results; (2) deceased donors with extended criteria; and (3) living related donors without genetic test results. Immunosuppressants such as low-dose calcineurin inhibitors or belatacept should be used, while mammalian target of rapamycin inhibitors should be avoided [18,31,32,34,36]. Anti-meningococcal vaccination, cytomegalovirus prophylaxis, influenza vaccination, and regular monitoring for infection should be performed (Fig. 3) [34,36].

Preventive strategy for thrombotic microangiopathy in kidney transplant (KT) recipients. Modified from Campistol et al. [36]. Modified from Zuber et al. [34], with permission from Elsevier. TMA, thrombotic microangiopathy; KTR, kidney transplant recipient; ESRD, end-stage renal disease; aHUS, atypical hemolytic uremic syndrome; MCP, membrane cofactor protein gene; CFH, complement factor H gene; CFI, complement factor I gene; CFB, complement factor B; anti-FH, anti-complement factor H antibodies; DSA, donor-specific antibody; CMV, cytomegalovirus; LTBI, latent tuberculosis infection; CNI, calcineurin inhibitors; mTOR, mammalian target of rapamycin.
Experts, including the Kidney Disease Improving Global Outcomes workgroup, recommend the prophylactic use of eculizumab in KT recipients with a high risk of recurrence based on genetic mutations [18,34,36]. For prophylactic use, eculizumab should be administered prior to surgery (i.e., on day 0 of transplantation) [18,31,34,80]. An additional dose was recommended on day 1 after surgery in some reports [80,87,88]. The use of eculizumab as a preemptive strategy in KT candidates with known aHUS is ongoing. Several studies have reported the clinical outcomes of KT recipients with eculizumab prophylaxis [89-91]. The results showed that eculizumab prophylaxis at the time of KT markedly reduced the incidence of recurrent TMA events, compared to data from the era before eculizumab [28,86]. In addition, eculizumab prophylaxis during preparation for KT resulted in better allograft outcomes than initiation of eculizumab following recurrence after KT. Emerging evidence suggests that eculizumab prophylaxis before KT is reasonable. However, plasmapheresis may be an alternative prophylactic strategy if eculizumab access is difficult [80,88,92].
Therapeutic strategies for recurrent aHUS in kidney transplantation
If any feature of TMA is observed after KT, early suspicion is important to improve clinical outcomes. Patients with post-transplant TMA exhibit different clinical features. In patients who exhibit only a change in renal function, allograft biopsy is necessary to confirm the diagnosis [31,37]. Therapeutic strategies such as plasmapheresis, immunosuppressant conversion, and rejection treatment should be implemented without delay [32]. Patients with post-transplant TMA showed an acceptable response to plasmapheresis in some studies [29,93,94]. However, patients who become dependent on or refractory to plasmapheresis should be considered for conversion to eculizumab [32,36]. Eculizumab contributed to the prevention and treatment of aHUS recurrence in KT recipients, whereas plasmapheresis was ineffective [95].
Treatment of pediatric aHUS
TTP is the first condition to be excluded in adult patients with probable aHUS; therefore, PE is typically applied until the ADAMTS13 activity result is reported. However, in children, TTP is uncommon and PE is not regarded as first-line treatment. Moreover, PE may induce complications related to central line catheter and extracorporeal circulation, particularly in young children (< 6 years of age). Therefore, PE should be applied only when necessary.
When primary aHUS and complement activation are suspected in children, the first-line treatment is administration of plasma to provide complement activation regulators that may be defective in the patient. Indeed, the treatment of choice is complement activation inhibitors, such as eculizumab. However, approval by Korean national health insurance is time-consuming, such that eculizumab may not be available immediately. Plasma infusion can ameliorate aHUS; therefore, it should be performed as a bridge treatment. When plasma infusion leads to volume overload or anti-CFH autoantibody is suspected, plasmapheresis is needed. When eculizumab becomes available, it should be administered immediately after vaccination for meningococcus, because untreated aHUS in children can be fatal; notably, delayed treatment results in kidney damage.
If a patient has a CFH mutation and experiences multiple relapses, liver transplantation can be considered because the majority of plasma CFH is produced by the liver. There have been several reports of successful liver transplantation that led to no further relapses [96]. While successful liver transplantation can cure CFH-associated recurrent aHUS, recipients of liver transplantation require immunosuppressive agents to prevent rejection, which leads to an immunocompromised state and an increased risk of infection and malignancy. These risks are in addition to medication-specific side effects such as growth impairment for steroids or post-transplant diabetes mellitus for calcineurin inhibitors. Therefore, the cost and risk of meningococcal infection during life-long treatment with eculizumab should be compared with the life-long side effects of liver transplantation. In addition, liver transplantation is applicable only for patients with CFH mutations, as well as those with CFI mutations, because the main source of CFH and CFI is the liver; for patients with mutations in other genes, liver transplantation is not available.
Key message
• Eculizumab is the first-line treatment for adult and pediatric patients with aHUS. If eculizumab is unavailable, PE and/or plasma infusion should be considered.
• KT recipients with a moderate-to-high risk of recurrence should receive prophylactic treatment with eculizumab. After KT, eculizumab should be considered for patients with TMA refractory to rejection therapy, immunosuppressant conversion, and plasmapheresis.
CONCLUSIONS
In this article, we defined the clinical diagnosis of TMA syndromes and proposed approaches for their differential diagnoses. The diagnosis of aHUS is complex and challenging, but is essential because of the devastating consequences of inadequate treatment. Advances in our understanding of the clinical, molecular, and functional aspects of the pathogenesis of aHUS are ongoing and have resulted in the development of novel therapeutic options. Several areas require further investigation, including factors that trigger aHUS and the optimal duration of treatment. Clinicians may refer to this current consensus for insights regarding diagnosis and treatment strategies for patients with aHUS.
Notes
No potential conflict of interest relevant to this article was reported.