Recent advances in the diagnosis and management of primary myelofibrosis
Article information
Abstract
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN) in which dysregulation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathways is the major pathogenic mechanism. Most patients with PMF carry a driver mutation in the JAK2, MPL (myeloproliferative leukemia), or CALR (calreticulin) genes. Mutations in epigenetic regulators and RNA splicing genes may also occur, and play critical roles in PMF disease progression. Based on revised World Health Organization diagnostic criteria for MPNs, both screening for driver mutations and bone marrow biopsy are required for a specific diagnosis. Clinical trials of JAK2 inhibitors for PMF have revealed significant efficacy for improving splenomegaly and constitutional symptoms. However, the currently available drug therapies for PMF do not improve survival. Although allogeneic stem cell transplantation is potentially curative, it is associated with substantial treatment-related morbidity and mortality. PMF is a heterogeneous disorder and decisions regarding treatments are often complicated, necessitating the use of prognostic models to determine the management of treatments for individual patients. This review focuses on the clinical aspects and outcomes of a cohort of Japanese patients with PMF, including discussion of recent advances in the management of PMF.
INTRODUCTION
Primary myelofibrosis (PMF), classified as a clonal myeloproliferative neoplasm (MPN), is characterized by the progressive proliferation of mainly granulocytic and megakaryocytic cells in the bone marrow. This condition leads to bone marrow fibrosis, resulting in subsequent extramedullary hematopoiesis and splenomegaly [1]. Typical clinical features of PMF include progressive anemia, symptomatic splenomegaly, and various constitutional symptoms [2-4]. Transformation to acute leukemia occurs in up to 20% of patients and this, along with progressive disease and infection, is one of the major causes of PMF-related death [2-4]. PMF is associated with a poor prognosis and a marked reduction in life expectancy [4-6], with median survival ranging from 3.5 to 6 years [7-11]. The molecular pathogenesis of PMF is characterized by dysregulation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathways, which are crucial for normal cytokine-mediated cell responses [12-14]. Approximately 85% of patients with PMF carry driver mutations in genes in these pathways [13], including JAK2V617F [15-18], MPLW515 [19,20], and calreticulin (CALR) [21,22]. In addition, there may be mutations in epigenetic regulators and RNA splicing genes that co-exist with driver mutations and play critical roles in disease progression [12,13]. In 2016, the World Health Organization revised the diagnostic criteria for MPN so that the diagnosis of PMF now requires screening for driver mutations in JAK2, CALR, and myeloproliferative leukemia (MPL) along with bone marrow biopsy (Table 1) [23]. If a patient has none of the driver mutations, which is a condition referred to as triple-negative disease, screening for non-driver mutations such as in additional sex combs like 1 (ASXL1), enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), tet methylcytosine dioxygenase 2 (TET2), isocitrate dehydrogenase 1/2 (IDH1/2), serine and arginine rich splicing factor 2 (SRSF2), and splicing factor 3b subunit 1 (SF3B1) is suggested. Another major revision was the identification of prefibrotic myelofibrosis as a new disease category separate from PMF and essential thrombocythemia (ET); distinguishing among these entities may have prognostic significance.

Current drug therapies for PMF, such as erythropoiesis-stimulating agents, hydroxyurea, and immunomodulatory drugs, do not improve survival. Although allogeneic stem cell transplantation (alloSCT) is potentially curative [3,24-26], it is associated with substantial treatment-related morbidity and mortality and is therefore only used in a minority of cases. Two phase III trials revealed that the JAK2 inhibitor ruxolitinib exhibits sustained efficacy and improves survival [27]. Decisions regarding the treatment of patients with PMF are often complicated, particularly with respect to the timing of alloSCT or the participation in clinical trials for novel drugs. Treatments are usually planned based on each patient’s estimated prognosis. Although several studies have addressed survival and prognostic factors in cohorts of patients with PMF, there is little such research in Asia to guide therapeutic decision-making. Accordingly, we started an annual nationwide survey of PMF cases in Japan in 1999, enrolling a total of 780 patients over a 17-year study period [28]. This review focuses on the clinical aspects and outcome of patients with PMF in the Japanese registry and discusses recent advances in the management of PMF.
CLINICAL FEATURES OF PMF
Clinical features at presentation
Most patients with PMF are aged > 60 years at initial diagnosis, and the reported median age at diagnosis ranges from 69 to 79 years [29]. The incidence of PMF in several registries ranges from 0.1 per 100,000 individuals per year to 1 per 100,000 per year [29,30]. The clinical manifestations of PMF vary. Most patients present with anemia, splenomegaly, and constitutional symptoms [2], but up to 30% of patients are asymptomatic at diagnosis. Splenomegaly can cause abdominal pain, abdominal discomfort, and early satiety. Constitutional symptoms include low-grade fever, night sweats, and weight loss. Some patients present with headache, inactivity, fatigue, insomnia, pruritus, bone pain, or thrombosis [31,32]; these symptoms significantly lower the patient’s quality of life (QOL). Various scales (e.g., European Organization for the Research and Treatment of Cancer [EORTC] QLQ-30, Functional Assessment of Cancer Therapy-Lymphoma [FACT-Lym], and the modified Myelofibrosis Symptom Assessment Form [MF-SAF]) are used to assess symptoms and QOL [33-35].
Driver mutation status and cytogenetic analysis
As noted above, most patients with PMF carry one of three mutually exclusive somatic driver mutations [13]; JAK2V617F in up to 60%, CALR in up to 20%, and MPL in up to 5% of patients. Additional non-driver mutations associated with epigenetic modification (TET2, ASXL1, EZH2, IDH1/2), RNA splicing (SRSF2, SF3B1, U2AF1 [U2 small nuclear RNA auxiliary factor 1]), JAK/STAT signaling (CBL [Casitas B-cell lymphoma], LNK), and DNA repair (TP53 [tumor protein p53]) are found in 1% to 10% of patients. In cytogenetic analyses, chromosomal abnormalities such as complex karyotypes and single or double abnormalities, including +8, del(7)/7q-, 12p-, inv(3), and 11q23 rearrangements, are defined as unfavorable karyotypes [36].
Clinical features of PMF in the Japanese registry cohort
As previously reported [28], questionnaires were sent annually to approximately 500 hematology departments of board-certified member institutes of the Japanese Society of Hematology. Patients newly diagnosed with PMF between 1999 and 2015 were entered into the registry and followed annually to collect information on outcome. The average response rate to the questionnaires was 48.0% (range, 44.7% to 49.7%). Approximately 50 patients were entered per year, yielding an eventual total of 780 patients with PMF in the cohort. The median follow-up at the time of analysis was 23 months and the median age at diagnosis was 66 years (range, 19 to 96) [28]. Low blood cell counts or other abnormal laboratory results were the most frequent reasons for initial consultation with a hematologist. Only 20% of patients initially presented with symptoms such as fatigue, weight loss, palpitations, shortness of breath, dizziness, abdominal fullness, fever, or abdominal pain. Splenomegaly was present in 75% of patients, and up to 70% had anemia, defined as a hemoglobin concentration < 10 g/dL. More than half of the patients had leukoerythroblastosis and a circulating blast frequency ≥ 1%. The clinical features, age distribution, and male-to-female ratios observed in our cohort were similar to those reported in European and North American studies [2,9,36,37]. Since items other than weight change for symptom assessment were not included in our registry data, symptom burden was not evaluated using recent scoring systems in our analysis. In our series, the JAK2V617F mutation was detected in 56% of the patients, but the MPL and CALR mutations were not found in most subjects. However, in a previously published study from our institution, the JAK2 mutation, an MPL exon 10 mutation, and a CALR exon 9 mutation were found in up to 60%, up to 2%, and 25% of the patients, respectively [38]. Ten percent of patients had no detectable mutations (i.e., triple-negative disease), and none had overlapping mutations. These results are consistent with data from larger cohort studies [12,13]. Bone marrow or blood karyotype analysis revealed abnormalities in 34% of cases. The most frequent abnormal karyotypes were del(20) and del(13). Unfavorable karyotypes were detected in 15% of the patients [28].
OUTCOME OF PMF
Survival
Long-term survival data in PMF patients were reported by the Mayo Clinic-Italian collaborative group, with a median survival of 5.9 years among 267 patients [39]. Patients with PMF had a worse survival than those with polycythemia vera (PV) (median survival, 13.5 years) or ET (median survival, 19.8 years). A similar result was reported by a Swedish group. In their population-based study, patients with PMF had higher relative survival ratios (RSRs), defined as the observed survival in the patient group divided by the expected survival of a comparable group from the general population, compared with those with PV and ET; the 10- and 20-year RSRs were 0.21 and 0.06 for PMF, 0.64 and 0.32 for PV, and 0.68 and 0.44 for ET, respectively [10].
Survival and causes of death in the Japanese registry cohort
In the Japanese registry cohort, the median survival was 47 months (95% confidence interval [CI], 41 to 53) at the time of the analysis. The 3-year overall survival (OS) was 59% (95% CI, 55% to 63%) (Fig. 1) [28]. These values are worse than those reported previously [7-11]. However, as the registry cohort was based on a questionnaire survey, it might have included patients with more advanced disease. We did not observe a plateau in survival curves during follow-up, indicating a persistent mortality rate that was attributable to PMF. Unfortunately, no improvement in survival was observed over the 17-year study period. Of the patients in the registry for whom the final cause of death was known, infection and leukemic transformation were the most frequent causes, followed by bleeding, PMF progression without transformation to leukemia, heart failure, and other malignancies (Table 2) [28].


ASSESSING PROGNOSIS IN PMF
Prognostic scoring systems
Several prognostic scoring systems have been established for PMF. The International Prognostic Scoring System (IPSS), developed in 2009, utilizes five independent risk factors at the time of diagnosis to predict survival: age > 65 years; hemoglobin level < 10 g/dL; leukocyte count > 25 × 109/L; circulating blast frequency ≥ 1%, and the presence of constitutional symptoms (Tables 3 and 4) [2]. The IPSS was subsequently refined to the Dynamic IPSS (DIPSS) in 2010 [37] and the DIPSS-plus in 2011 [36]. The DIPSS-plus incorporates information such as an unfavorable karyotype, need for red cell transfusion, and platelet count < 100 × 109/L, and stratifies patients into four risk groups: low (no risk factors); intermediate-1 (one risk factor); intermediate-2 (two or three risk factors); and high (four or more risk factors), with median OS of 185, 78, 35, and 16 months, respectively. The advantage of DIPSS and DIPSS-plus is that they can be applied to patients at any time during the disease course. These scoring systems are based on easily assessable clinical characteristics and blood counts, and are currently the standard methods used to estimate the prognosis of individual patients.

International prognostic scoring systems for primary myelofibrosis

Prognosis of primary myelofibrosis according to international prognostic scoring systems
More recently, a number of genetic risk factors independent of DIPSS-plus were identified, including driver gene mutational status [39-42], JAK2 allele burden [43], and the presence or number of other non-driver somatic mutations [44,45]. In most patients with PMF with one of the abovementioned three driver mutations, Tefferi et al. [40] showed that the CALR mutation was associated with a better outcome than the other two driver mutations, independent of the DIPSS-plus estimated risk. In addition, patients with triple-negative disease had a much higher incidence of leukemic transformation and an unfavorable outcome. As for non-driver mutations, the presence of mutations in ASXL1, EZH2, SRSF2, or IDH1/2 is defined as high molecular risk, as these mutations are associated with poor prognosis and risk of leukemic transformation [44]. The number of these detrimental mutations is also an adverse risk factor, and the presence of two or more non-driver mutations predicts the worst survival [45]. Notably, these molecular risk factors predict a poor outcome independent of conventional prognostic scoring systems.
Stratification according to PMF prognostic scoring in the Japanese registry cohort
The registry cohort was divided into four prognostic groups according to the various prognostic models reported previously (Fig. 2) [28]. Both IPSS and DIPSS differentiated between low and intermediate-1 risk patients, but could not distinguish high risk from intermediate-2 risk. In contrast, the DIPSS-plus model accurately divided the population into four statistically different risk groups. The inclusion of the need for red cell transfusion and an unfavorable karyotype are likely the factors that account for the good performance of DIPSS-plus, as these were the most significant predictors of shorter survival in our study [28]. Therefore, DIPSS-plus appears to be the optimal model for predicting survival in Japanese patients with PMF, although IPSS and DIPSS could also distinguish patients at higher versus lower risk of mortality. Moreover, the DIPSS-plus model could successfully identify patients with a poor prognosis at any time during the clinical course. This makes it useful for decision-making, for example when considering alloSCT. However, it had only modest accuracy for prognostication, suggesting that covariates included in DIPSS-plus alone are not sufficient, and that information regarding gene mutation status (e.g., CALR and ASXL1) should be included. We did not have data on mutations other than JAK2V617F in the registry. It is essential to collect precise genetic information to improve prognostication for patients with PMF throughout the disease course and subsequently inform the best treatment options.

Survival curves of 780 patients with primary myelofibrosis in a Japanese registry, stratified by prognostic scoring system risk groups at diagnosis. (A) International Prognostic Scoring System (IPSS), (B) Dynamic IPSS (DIPSS), (C) Dynamic IPSS plus additional prognostic factors (DIPSS plus). Adapted from Takenaka et al., with permission from Springer Nature [28].
TREATMENT OF PMF
Management of PMF
As noted above, the prognosis for patients with PMF is dismal compared with other MPNs, and alloSCT is the only current potentially curative treatment. However, as the median age at onset of PMF is 66 years, very few patients are eligible for alloSCT. For those who are not, therapy is primarily symptomatic, such as treating anemia or splenomegaly [3,4,11,26]. Treatment should be determined on a case-by-case risk assessment and evaluation of QOL using the DIPSS-plus risk classification and MF-SAF (Fig. 3). In terms of DIPSS-plus risk, a “watch-and-wait” policy is advisable for asymptomatic patients in the low and intermediate-1 risk groups because longterm survival can be expected with supportive therapy alone. For patients with symptomatic anemia, splenomegaly, or moderate constitutional symptoms, specific treatment is aimed at each particular symptom. PMF-associated anemia is usually treated with androgens, prednisolone, and danazol, which achieves improvement in up to 15% to 25% of patients [46,47]. Hydroxyurea is the first-line therapy for symptomatic splenomegaly, yielding an overall response rate of up to 40% and lasting an average of 1 year [47]. Recently, JAK2 inhibitors have been used for splenomegaly instead of hydroxyurea (see below). Splenectomy is an alternative choice for symptomatic splenomegaly, but it has a perioperative mortality rate of up to 5% to 10% and a morbidity rate of 25% [48,49]. Splenic irradiation can be used for patients who are not candidates for a JAK2 inhibitor or splenectomy, but the response is transient and there is a risk of severe cytopenia [50]. For patients in the intermediate-2 and high-risk groups, alloSCT should be considered if an appropriate donor exists. For those not eligible for alloSCT, a JAK2 inhibitor or participation in a clinical trial can be considered.
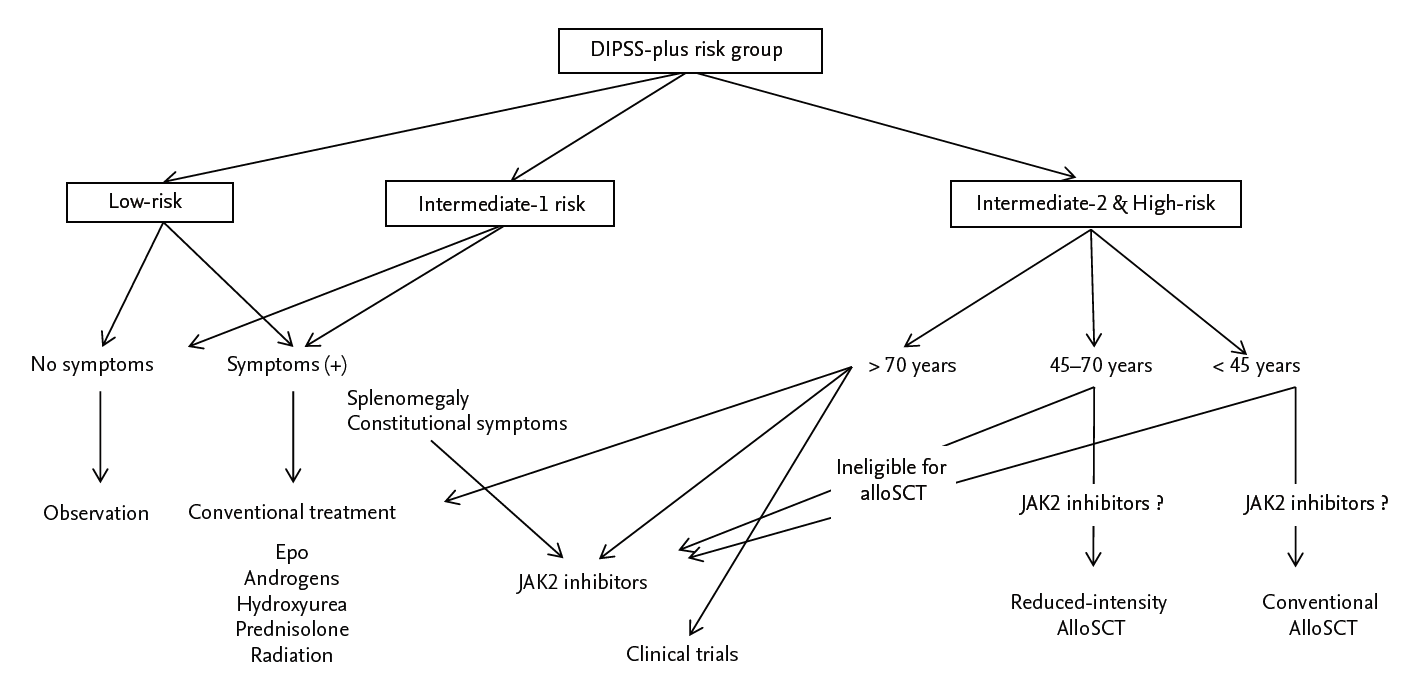
Treatment algorithm for primary myelofibrosis. DIPSS plus, Dynamic International Prognostic Scoring System plus additional prognostic factors; epo, erythropoietin; alloSCT, allogeneic stem cell transplant; JAK2, Janus kinase 2.
JAK2 inhibitors
The JAK2 inhibitor ruxolitinib has been available in the United States for the treatment of intermediate-2 or high-risk PMF since 2011. Two randomized phase III studies in the United States (Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment I [COMFORT-1] trial) and Europe (COMFORT-2 trial) were conducted for patients in the intermediate-2 or high risk groups with PMF or post PV/ET myelofibrosis, with trial results published in 2012 [33,51]. In the COMFORT-1 trial, 309 patients were assigned to either ruxolitinib or placebo. The COMFORT-2 trial included 219 patients randomized to receive either ruxolitinib or the best available therapy. Ruxolitinib administration improved splenomegaly and general symptoms such as fever, malaise, weight loss, and decreased activity. In 3 years of follow-up reports, the reduction in spleen volume and improvement in QOL were maintained, and improved survival was also observed [27,52]. These benefits were achieved regardless of the JAK2 mutation status, because the JAK2 inhibitor effectively suppresses proinflammatory cytokines, which is a major factor involved in the progression of PMF. Recently, the results of a pooled analysis of OS in COMFORT-1 and -2 were reported after crossover from the control groups to the ruxolitinib group in both studies; most patients in the control groups crossed over to ruxolitinib [53]. The OS at 144 weeks was 78% in the ruxolitinib group, 61% in the intention-to-treat control group, and 31% in the control group corrected for crossover, confirming the survival advantage conferred by ruxolitinib compared with placebo or other treatment. Spleen size at baseline was a risk factor for poor OS, and reduction in spleen size with ruxolitinib correlated with longer survival. The major adverse events were anemia and thrombocytopenia. Most of the grade 3 and 4 cytopenias appeared within 6 months (especially the first 2 to 3 months) of the start of treatment. Sudden interruption or withdrawal of JAK2 inhibitor provoked acute symptoms due to cytokine release syndrome; therefore, the dose of ruxolitinib should be tapered over up to 7 to 10 days to avoid this withdrawal syndrome. Since ruxolitinib inhibits T-cell function, attention should also be paid to the possible development of opportunistic infections, including tuberculosis, reactivation of hepatitis B, herpes zoster, or urinary tract infection [54]. Even if a patient is eligible for alloSCT, pre-transplant therapy with JAK2 inhibitor may be beneficial as splenomegaly has a negative impact on engraftment and transplant outcomes. A poor performance status due to constitutional symptoms increases non-relapse mortality after transplant. Therefore, pre-transplant ruxolitinib may improve performance status, reduce splenomegaly, and decrease the grade of subsequent acute graft-versus-host disease, resulting in faster hematopoietic recovery, improved graft function, and improved survival. However, the use of ruxolitinib before alloSCT also has potentially detrimental effects, including withdrawal syndrome when the drug is discontinued, increased risk of infections, delayed hematopoietic recovery, and potential tumor lysis syndrome. As clinical experience is limited at present, use of a JAK2 inhibitor before alloSCT should be limited to patients enrolled in clinical trials.
AlloSCT
A retrospective analysis using the Center for International Bone Marrow Transplant Research database assessed transplant outcomes for 289 patients (median age, 47 years) with PMF [55]. The 5-year disease-free survival and 100-day treatment-related mortality rates were 33% and 18% for human leukocyte antigen (HLA) identical sibling donor transplants, 27% and 35% for unrelated donor transplants, and 22% and 19% for transplants from alternative related donors, respectively. Splenectomy was performed in 65 patients prior to transplantation but was not associated with better survival. Further research indicated that alloSCT can engraft despite severe bone marrow fibrosis, with an incidence of graft failure of < 10%; long-term survival is achieved in up to 30% to 50% of patients [56]. It was also reported that bone marrow fibrosis disappeared in more than half of patients with sustained engraftment. However, the incidence of transplant-related mortality may be as high as 30% to 50%. Risk factors for transplant-related mortality include huge splenomegaly, the need for frequent red blood cell transfusions, transplantation from HLA-mismatched donors, low performance status, and a high comorbidity index [57].
Recently, reduced-intensity conditioning regimens have shown promise, typically for the elderly and those with comorbidities. While such regimens are feasible, they do not appear to improve outcome [58-63]. No randomized controlled trials have compared myeloablative and reduced-intensity conditioning for alloSCT. Therefore, the optimal intensity of the conditioning regimen still needs to be defined. An alloSCT from a haploidentical related donor is also a consideration when HLA-identical donors are not available, but the results of haploidentical donor transplantation should be evaluated in clinical trials. A consensus report from an international working group suggested that patients aged < 70 years with intermediate-2 or high-risk PMF are candidates for alloSCT. In addition, they recommend that alloSCT be considered even for patients with intermediate-1 risk disease who are aged < 65 years, if they have transfusion-dependent anemia, > 2% peripheral blood blasts, or adverse cytogenetics [57]. There is still much interest in the use of JAK2 inhibitors before alloSCT, and several clinical trials are ongoing to evaluate the efficacy and safety of JAK2 inhibitors for this purpose [41,64].
Treatment in the Japanese registry cohort
Red cell transfusions, androgens, hydroxyurea, and ruxolitinib were administered to 192 (25%), 72 (9%), 83 (11%), and 47 patients (6%), respectively [28]. Eleven patients (1%) underwent splenectomy and 21 (3%) had splenic radiation. An additional 43 patients (6%) received alloSCT at a median of 343 days (range, 23 to 4,066) after diagnosis and a median age of 52 years (range, 24 to 66). The stem cell sources for a first alloSCT included unrelated donor bone marrow (n = 21), related donor peripheral blood stem cells (n = 14), cord blood (n = 5), and related donor bone marrow (n = 2). Six patients received a second allo-SCT for disease relapse, and one patient received a third alloSCT. The median follow-up after the first alloSCT was 36 months (range, 3 to 130), and the 3-year OS was 84% (95% CI, 68% to 93%). The estimated median survival after the first alloSCT was 134 months (range, 71 to not reached), and the median survival after diagnosis among all patients who underwent alloSCT was 207 months (range, 105 to not reached). The latter value is significantly longer than that calculated for patients who did not undergo alloSCT (median, 45 months; range, 38 to 49; p < 0.001). Although ours was not a controlled trial and the patients undergoing alloSCT were younger than those who did not, the registry data suggest that alloSCT could prolong OS and thereby affect the natural course of PMF. In contrast, no improvement in OS was observed in the 47 patients treated with a JAK2 inhibitor in our series, although this was likely due to the small number of patients and the short observation period from the introduction of the agent.
CONCLUSIONS
Based on recent advances in our understanding of the molecular basis of PMF and the revised diagnostic criteria for MPN, screening for the JAK2, CALR, and MPL driver mutations and assessing bone marrow morphology have become essential for the diagnosis of PMF. Several prognostic scoring systems have been developed for PMF based on clinical characteristics and blood counts. While these are the standard methods currently used to estimate prognosis, it appears that evaluating other molecular risk predictors may improve the assessment of individual patients independent of the conventional prognostic scoring system. Choice of treatment should be based on risk assessment and the evaluation of the QOL in each case, generally using the DIPSS-plus risk classification and MF-SAF. The JAK2 inhibitor ruxolitinib can reduce spleen volume, and improve QOL and survival. However, alloSCT is currently the only potentially curative treatment for PMF, although it continues to have high transplant-related mortality. Ongoing trials of JAK2 inhibitors before alloSCT may help clarify the value of this approach as we continue to search for the best options for the treatment of PMF.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was performed with the support of the Idiopathic Disorders of Hematopoietic Organs Research Committee of Japan. The authors are grateful to the collaborators for their valuable contributions and acknowledge all 291 institutions that participated in the present study.