How to tackle congestion in acute heart failure
Article information
Abstract
Acute heart failure is a common complication of chronic heart failure and is associated with a high risk for subsequent mortality and morbidity. In 90% of case acute heart failure is the resultant of congestion, a manifestation of fluid build-up due to increased filling pressures. As residual congestion at discharge following an acute heart failure episodes is one of the strongest predictors of poor outcome, the goal of therapy should be to resolve congestion completely. Important to comprehend is that increased cardiovascular filling pressures are not solely the resultant of intravascular volume excess but can also be induced by a decreased venous capacitance. This review article focusses on the pathophysiology, diagnoses, and treatment of congestion in acute heart failure. A clear distinction is made between states of volume overload (intravascular volume excess) or volume redistribution (decreased venous capacitance) contributing to congestion in acute heart failure.
INTRODUCTION
Acute heart failure (AHF) represents a seminal event in the disease trajectory of chronic heart failure (CHF) [1] and is associated with increased risks of re-hospitalization and mortality [2]. The costs of frequent readmissions to the hospital create a significant economic burden on society [3]. In total, 90% of AHF cases that require hospitalization are due to worsening congestion, and only a minority of these cases are due to low cardiac output [4]. Many drugs have been developed in the last decade to reduce the burden of congestion during AHF [5-10]. However, numerous trials have failed to significantly decrease the risks of hospitalization for heart failure and all-cause mortality, suggesting that it is difficult to influence long-term outcomes using transitory medication. Many trials have also indicated that congestion is often unresolved when patients are discharged. AHF therapies have not changed dramatically in the past few years, and diuretics and vasodilators remain the mainstays of treatment [11,12]. Although cardiologists have confidence in the efficacy of diuretics and vasodilators, there is a lack of information on how to titrate these therapies [11,12].
This review provides an overview of the pathophysiology of congestion in heart failure, focusing on effective strategies for alleviating congestion. It emphasizes the importance of achieving complete decongestion, because residual congestion is the most significant predictor of hospital readmission and postdischarge mortality [13].
UNDERSTANDING CONGESTION
What is congestion?
Congestion is defined as the signs and symptoms of extracellular fluid accumulation, instigated by an increase in left-sided cardiac filling pressure [14]. This definition recognizes that poor cardiac function is a prerequisite for developing congestion. However, numerous other organs play a role in the development of congestion, including the splanchnic veins and the interstitial and endothelial tissues. The use of implantable hemodynamic monitoring devices have significantly contributed to our understanding of how CHF develops into AHF [15]. One of the earliest changes is a small pressure increase in the pulmonary artery or right side of the heart, which takes place over an extended time period. This often occurs weeks in advance of the patient presenting at a hospital with AHF. Once filling pressures significantly increase, symptoms of functional deterioration occur that often result in patients going to the emergency department. This process is often described as the transition between hemodynamic congestion and clinical congestion. At this stage, clinical congestion might lead to pulmonary edema with desaturation, myocardial ischemia, a progressive decline in kidney function, cerebral changes, and an increased risk of arrhythmia. In this way, a disease that normally progresses slowly is transformed into a life-threatening condition.
What mechanisms produce congestion?
The progressive accumulation of sodium and water is often considered the major reason why clinical congestion leads to AHF. Indeed, heart failure is frequently characterized as compensatory neurohormonal activation that leads to chronic sodium and water retention. Alterations in both the proximal and distal parts of the nephron play a central role in the enhanced avidity for sodium that occurs in the kidneys during heart failure [16]. Approximately 65% of this retained sodium is stored in the extracellular compartment [17], which consists of both an intravascular compartment and the interstitium. Sodium is retained iso-osmotically, with approximately 75% retained in the interstitium and only 25% retained in the intravascular compartment. Therefore, changes in total blood volume are always associated with a 3-fold increase in interstitial volume. In addition, changes in total blood volume are not evenly distributed across the arterial and venous systems. The venous system contains up to 70% of the total blood volume, most of which is in the high capacitance splanchnic veins [18]. These splanchnic veins can accommodate a 65% increase in blood volume without changes in central filling pressures [19]. Therefore, a patient’s weight must have increased significantly in the weeks prior to hospitalization for heart failure, if central filling pressures have increased solely by iso-osmotic sodium retention. Indeed, studies on patients who present with AHF and signs of weight gain have shown that the blood-volume compartment expands by approximately 40%. Assuming an average blood volume of 5 L, this would mean an expansion of 1.9 L. However, because approximately 75% of the sodium is retained in the interstitium, there is an additive retention of 5.85 L in the interstitium [20]. Follow-up studies on these patients with decongestion indicated that most of the diuresed volume originated in the interstitium. By contrast, several studies have found that nearly 50% of patients presenting with AHF did not have significant weight gain prior to decompensation [21]. Therefore, chronic progressive volume expansion is not the only reason for congestion.
Changes in the capacitance of the splanchnic veins can also contribute to the development of congestion [22]. The splanchnic system can act as a volume buffer to protect the cardiovascular system from blood loss. By decreasing their capacitance, mediated by activation of the sympathetic nervous system, the splanchnic veins can augment their cardiac preload and maintain cardiac output when blood volume is low [23]. However, these changes in venous capacitance probably also contribute to the development of congestion, particularly in patients who have not gained weight [24,25]. Therefore, both changes in total blood volume (volume overload) and venous capacitance (volume redistribution) can lead to congestion. Indeed, as of 2016, the European Society Guidelines for the treatment of acute and CHF recommend that clinicians differentiate between volume overload and volume redistribution when treating AHF. However, it is clear that the two mechanisms are not mutually exclusive and both may contribute to congestion in many patients with AHF (Fig. 1).
Relationship between intravascular volume and filling pressures in acute heart failure.
The importance of resolving congestion completely
Irrespective of the mechanisms causing congestion, once it has developed, congestion is self-perpetuated in a variety of ways. Studies of selectively induced coronary-sinus congestion in dogs demonstrated a decrease in coronary perfusion and changes in myocardial ventricular stiffening with a downward and rightward shift in the end-systolic pressure volume relationship [26-28]. Changes in central venous pressure are also associated with worsening renal function (WRF), resulting in a drop in natriuresis [29-31]. Heart-rate variability data showed a shift from autonomic toward sympathetic nervous system control [32], which can further decrease splanchnic venous capacitance and increase preload [22]. In addition, splanchnic congestion results in increased intra-abdominal pressure, WRF, and ischemia [22,33]. Ischemia reduces the effectiveness of the intestinal barrier, leading to lipopolysaccharide-induced endotoxicity and increasing inflammation [22]. Colombo et al. [34] used a selective forearm-congestion model to demonstrate that transient congestion results in endothelial activation and promotes inflammation. The ensuing endothelial activation results in decreased arterial impedance and venous capacitance, which negatively influences afterload and preload in AHF. Therefore, congestion is self-amplifying, and complete decongestion should always be the goal when treating patients with this condition. It is not surprising that achieving decongestion in patients with AHF or those awaiting transplants for advanced heart failure is associated with improved outcomes. Indeed, residual congestion at discharge following hospitalization for AHF is the strongest predictor of subsequent readmission and cardiovascular mortality.
EVALUATION OF THE CONGESTED PATIENT
Is the patient congested?
Determining the presence of clinical congestion is often easy for the experienced physician. Clinical symptoms and signs of dyspnea, orthopnea, bendopnea, rales, the third heart sound, jugular venous distention, and pulmonary or peripheral edema all point to congestion. When combined, these signs have high specificity for detecting increased cardiac filling pressures. However, even a combination of rales, elevated jugular venous distention, and edema only has a 58% sensitivity for detecting an elevated pulmonary capillary wedge pressure. Therefore, auxiliary tests are often necessary to detect hemodynamic congestion, which lacks clear clinical signs or symptoms. The gold standard for assessing congestion remains right heart catheterization and direct measurements of right atrial and pulmonary capillary wedge pressure using a pulmonary artery catheter [35]. In most patients with heart failure, the right-sided and left-sided filling pressures are closely related [36]. Therefore, right-sided measurements can be used as a barometer for left-sided pressure in most patients. The Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial failed to demonstrate that direct measurements of filling pressures with tailored therapy and a pulmonary artery catheter were superior to clinically guided assessments of AHF patients [35]. As a result, the use of invasive tailored therapy to treat AHF has declined. However, a pulmonary artery catheter remains useful for patients with unclear hemodynamic profiles at baseline or uncertain hemodynamic responses following therapy (e.g., WRF, right-left mismatches, or worsening heart failure). Cardiac hemodynamics are now frequently assessed by non-invasive methods. Echocardiography provides a comprehensive non-invasive evaluation of cardiac hemodynamics [37]. Tables 1 and 2 provides an overview of the echo-derived parameters used to asses right-sided versus left-sided filling pressure. These echocardiographic parameters can also detect subclinical congestion. Some data have suggested that targeted decongestion, based on echocardiography, can reduce the risk for hospitalization for heart failure [38]. In addition to echocardiography, natriuretic peptides (NPs) are increasingly being used to assess cardiac filling pressures non-invasively, and are useful for diagnosing the presence of congestion and stratifying patients according to risk. However, their value in determining the success of decongestive therapy remains questionable [39]. Indeed, NP levels probably reflect atrial dimensions and are affected by changes in local geometry and stretch triggers, which are influenced by age, body mass index, and kidney function [39]. An additional problem is the lag times in changes in NP levels following hemodynamic changes, as these levels equilibrate approximately 1 week after clinical stabilization [39].

Right-sided assessment: sensitivity and specificity for variables predicting right atrial pressure > 7 mmHg
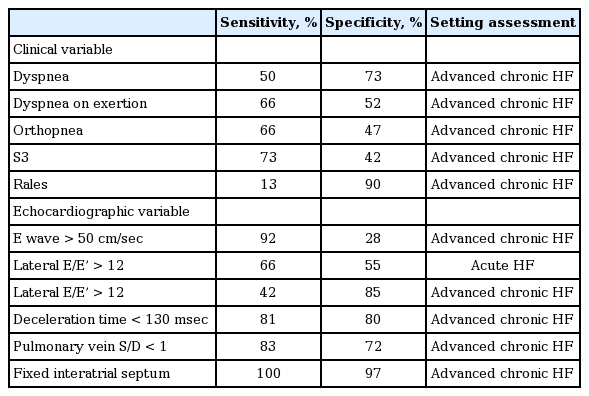
Left-sided assessment: sensitivity and specificity for variables predicting pulmonary capillary wedge pressure > 18 mmHg
Does volume overload or volume redistribution predominate?
Filling pressures are poor surrogates for intravascular volume because they are also affected by venous compliance. A meta-analysis of studies comparing central venous pressure with blood volume measurements indicated that only 2.5% of central venous pressure variability across the study population was explained by differences in blood volume. Therefore, the observation that nearly 50% of AHF patients had gained less than 1 kg in weight is unsurprising, because both volume overload (changes in volume) and volume redistribution (changes in venous compliance) can contribute to congestion. At present, no study has evaluated clinical or technical strategies for differentiating between these causes of AHF. However, significant weight gain during the week prior hospitalization for heart failure and peripheral edema suggests that expansion of the extracellular compartment, and therefore volume overload, has occurred. More recently, several AHF studies have examined the feasibility of using direct plasma volume measurements to address this problem. Miller and Mullan [20] measured the blood volume of 26 AHF patients with a clinical diagnosis of volume overload. The measurements showed that the plasma-volume compartment expanded to different degrees among AHF patients. However, 8% of the patients with a clinical diagnosis of volume overload actually had decreased plasma volumes [20] and may have had volume misdistribution rather than volume overload. These findings indicate that directly measuring plasma volume may be useful for differentiating among causes of congestion and for titrating therapies. However, at present, a detailed patient history that includes weight changes and clinical assessments of edema (e.g., peripheral edema, ascites, bowel edema) may be the most reliable method for differentiating between volume overload and redistribution [40].
TREATMENT OF CONGESTION
Because one of the strongest predictors of re-hospitalization and mortality in AHF patients is residual congestion on discharge from hospital, the goal of therapy should be to achieve complete decongestion [13]. In addition, once congestion has been resolved, it is important to prevent it from recurring. Although volume overload and volume redistribution can overlap in clinical practice, we propose that the emphasis of decongestive therapy should be on the prevailing mechanism of congestion.
Tackling congestion with volume overload
When volume overload is causing congestion, the goal of therapy should be to decrease extracellular sodium and water, bearing in mind that extracellular volume is found in both the interstitium and plasma [17]. In addition, the only physiological methods of removing excessive sodium and water are kidney-mediated natriuresis and diuresis [16]. Direct diuresis of the interstitium is not possible, and to decongest the interstitium, the Starling forces between the interstitium and the plasma compartment need to be reversed [41]. This is called ‘plasma-refilling’ and is achieved by decreasing hydrostatic capillary pressure and increasing oncotic capillary pressure of the plasma compartment, secondary to renal natriuresis and diuresis. To effectively tackle volume overload, a thorough understanding of the renal changes that cause increased sodium avidity in AHF patients is necessary. This information is beyond the scope of the present review, but has previously been described [16]. Loop diuretics remain the mainstay of decongestive therapy for volume overload. However, if diuretic efficacy is low, combinational diuretic therapy may be necessary to enhance the diuretic response and facilitate decongestion (Fig. 2) [42].

Targets for combinational diuretic therapy in volume overload. QD, once a day; HCTZ, hydrochlorothiazide; eGFR, estimated glomerular filtration rate; BUN, blood urea nitrogen; SGLT-2, sodium-glucose-linked transporter 2; MRA, mineralocorticoid receptor antagonist; HF, heart failure.
Loop diuretics
Loop diuretics are the mainstay of diuretic therapy for AHF, and almost 90% of patients in the ADHERE (Acute Decompensated Heart Failure National Registry) database were treated using intravenous loop diuretics [43]. In addition, in 63% of patients, loop diuretics are the sole drug therapy used to combat AHF [43]. Despite the ubiquitous use of loop diuretics, only one randomized controlled trial, the Diuretic Optimization Strategies Evaluation (DOSE) trial, has prospectively assessed their performance in treating AHF [44]. In the DOSE trial, 308 patients were randomly assigned low and high doses of diuretics (2 × 2), either continuously or in bolus form. No differences were observed between patients who had diuretics administered continuously compared to those who ingested them in bolus form. However, those patients receiving the higher dose of furosemide (median dose of 773 mg vs. 358 mg in 72 hours) showed a trend towards faster relief from dyspnea and significantly higher net fluid and weight losses. When the glomerular filtration rate is low, it is often necessary to increase the dose of loop diuretics. Recent research has also shown that early interventions using loop diuretics may be associated with improved outcomes in AHF patients [45]. However, treatment with loop diuretics may also be associated with neurohormonal activation and ionic disturbances and has never been shown to reduce mortality rates [46]. In addition, a significant number of patients treated with loop diuretics do not achieve decongestion, and it is unclear whether these patients would benefit from an increased dose or combination-diuretic therapy.
Combination-diuretic treatment and bail-out therapy
If loop diuretics fail to relieve volume overload, there is little information to suggest the best alternative treatment. Indeed, changing the dose or using another loop diuretic are frequently suggested by practice guidelines [12]. Fig. 2 provides an overview of the targets within the kidney nephron and the potential strategies for enhancing natriuresis and diuresis. Thiazide diuretics are frequently used to treat patients who are resistant to loop diuretics [47]. Indeed, prolonged used of loop diuretics can result in intrinsic renal adaptations, including enhanced sodium avidity, which are largely mediated by thiazide-sensitive sodium chloride transporters [48]. One limitation of thiazides is that they reduce the kidneys’ capacity for producing dilute urine. Therefore, they should be avoided by patients with hypotonic hyponatremia [49]. Mineralocorticoid receptor antagonists are an intrinsic part of CHF therapy; however, at higher doses they have natriuretic effects [50]. They are often used when loop diuretics induce renal potassium wasting. The recent ATHENA-HF (Aldosterone Targeted Neurohormonal Combined with Natriuresis Therapy in Heart Failure) trial failed to show that a high dose of spironolactone improved decongestion when administered with standard therapy and evaluated using changes in NP levels at 96 hours, compared to placebo [51]. Acetazolamide is an outdated diuretic that is still used to treat mountain sickness and glaucoma. One observational study found that when acetazolamide was used along with loop diuretics to treat AHF, the diuretic efficacy increased by 100 mg of sodium per 40 mg furosemide equivalents [52]. Currently, one small prospective study (NCT01973335) is evaluating the effects of acetazolamide in AHF patients. Sodium-glucose-linked transporter 2 (SGLT-2) inhibitors also induce natriuresis and reduce plasma volume by inhibiting proximal nephron sodium reabsorption [53]. Two trials of diabetic patients with cardiovascular disease showed that SGLT-2 inhibitors were highly effective for decreasing the likelihood of hospitalization for heart failure [54,55]. Empagliflozine is currently being tested in stable heart failure patients with both reduced (NCT03057977) and preserved (NCT03057951) ejection fractions. However, its potential for treating patients with AHF is unclear. If congestion persists despite therapy with diuretics, bail-out therapy with ultrafiltration may be necessary. The use of ultrafiltration to treat AHF patients with persistent congestion is beyond the scope of this review, but has recently been reviewed in detail [56].
When is volume overload completely resolved?
One of the biggest problems in treating patients who have congestion with volume overload is knowing when euvolemia has been achieved. Retrospective analyses of the DOSE and CARESS (Clopidogrel and Aspirin for Reduction of Emboli in Symptomatic Carotid Stenosis) trials have indicated that even in patients with no edema or orthopnea and a jugular venous pressure < 8 mmHg at discharge, the risk for readmission, death, or unplanned hospital visits within 60 days was 50% [57]. This suggests that clinical assessments are insufficient to detect euvolemia. A retrospective analysis of the CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Patients (CHAMPION) trial indicated that patients in the clinical arm were deemed optivolemic at much higher filling pressures than patients in the hemodynamic guided arm, inevitably resulting in higher readmission rates [58]. Testani et al. [59] showed that patients who achieved hemoconcentration during AHF treatment had significantly better prognoses, even if WRF developed [59-62]. Hemoconcentration occurs when both the interstitium and plasma compartment are free of excess volume, and additional diuresis will not result in the interstitium refilling the plasma compartment [61]. However, whether hemoconcentration can be used to guide decongestion has not been tested, and the intrinsic fluctuations in hemoglobin concentrations may be problematic. Further studies are needed to determine when a congested patient with volume overload is dry [63].
Tackling congestion with volume redistribution
When volume redistribution causes congestion, therapy aims to enhance venous capacitance function and lower cardiac filling pressures [64]. To achieve this, a combination of vasodilators and low doses of intravenous diuretics are used. Intravenous vasodilators reduce preload by venodilation; thereby, enhancing venous capacitance; they also induce arteriolar vasodilation, which reduces the afterload on the failing heart [65], allowing the heart to activate the Frank-Starling mechanism with a drop in left ventricular end-diastolic pressure and a parallel increase in stroke volume. Initial bed rest can also enhance the effects of intravenous dilatory drugs [66].
Central filling pressures often drop spontaneously following right heart catheterization and the patient’s admission to a cardiac care unit, even before any vasoactive drugs have been administered [50]. This may be due to a reduction in adrenergic stress, which enhances venous compliance. Although the ESCAPE trial did not demonstrate the benefit of targeting filling pressures (goal: right atrial pressure < 8 mmHg; pulmonary capillary wedge pressure < 15 mmHg), lower filling pressures in patients awaiting heart transplants are associated with improved outcomes [35,67,68]. Similar findings were documented in the CHAMPION trial [58]. Numerous vasodilatory agents are being developed that improve venous compliance and capacitance and target systemic vascular resistance, with secondary improvements in cardiac output and pulmonary capillary wedge pressure. These novel drugs include cenderitide, β-arrestin-biased angiotensin II type 1-receptor blockers, nitroxyl-donors, soluble guanylate cyclase modulators, and nicorandil [69]. However, several recently developed vasodilators failed to improve clinical outcome in heart failure patients [10,70]. Once filling pressures have been normalized, tapering of intravenous vasodilatory drugs should be accompanied by uptitration of neurohumoral blockers [71]. Refractory or recurrent volume misdistribution is often caused by poor intrinsic cardiac function. Increased cardiac filling pressures activate the venous system to meet the preload demand. Patients should be meticulously evaluated to determine their suitability for advanced therapies including cardiac resynchronization therapy, left ventricular assist devices, and heart transplants [64]. Because the risk of recurrence is related to the total duration of high filling pressures, monitoring filling pressures in these patients is particularly important [72].
Preventing recurrence of congestion
In addition to achieving decongestion, one of the most effective interventions is an appropriate discharge policy. Titration of neurohormonal blockers is essential. These include β-blockers, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, and angiotensin receptor/neprilysin inhibitors. The discharge policy should ensure that ambulatory rehabilitation is initiated and a low salt diet with fluid restriction is implemented. In addition, a stable dose of oral diuretics must be formulated, and follow-up procedures should be in place to monitor the patient [73]. In the OPTIMIZE-HF (Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure) trial, treatments with β-blockers and ACE-inhibitors were strongly associated with better postdischarge outcomes [74]. A recent analysis also found that achieving decongestion and uptitrating ACE-inhibition has a synergistic effect [75], suggesting that therapies should be optimized while treatment for decongestion is ongoing.
CONCLUSIONS
Improving our understanding of the pathophysiology of congestion allows us to explain why congestion at discharge results in a poor prognosis for AHF patients. Increased filling pressures are not always associated with volume excesses. A careful evaluation of the congested patient is necessary to determine whether volume overload or volume misdistribution is primarily responsible for congestion. In both cases, achieving complete decongestion is the target. If volume overload is present, well-designed diuretic therapies may be necessary. In patients with volume misdistribution, a combination of vasodilators and low-dose diuretics may be implemented. Once decongestion has been achieved, every effort should be made to prevent congestion recurring. A well-designed heart-failure-care program, emphasizing neurohormonal-blocker uptitration, rehabilitation, follow-up procedures, the treatment of comorbidities, device-based therapies, sodium restriction, and education can reduce the risk for recurrence.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
Pieter Martens is supported by a doctoral fellowship by the Research Foundation-Flanders (FWO, grant-number: 1127917N). Pieter Martens and Wilfried Mullens are researchers for the Limburg Clinical Research Program (LCRP) UHasselt-ZOL-Jessa, supported by the foundation Limburg Sterk Merk (LSM), Hasselt University, Ziekenhuis Oost-Limburg and Jessa Hospital.