Infectious complications after hematopoietic stem cell transplantation: current status and future perspectives in Korea
Article information
Abstract
Hematopoietic stem cell transplantation (HSCT) is a treatment for hematologic malignancies, immune deficiencies, or genetic diseases, ect. Recently, the number of HSCTs performed in Korea has increased and the outcomes have improved. However, infectious complications account for most of the morbidity and mortality after HSCT. Post-HSCT infectious complications are usually classified according to the time after HSCT: pre-engraftment, immediate post-engraftment, and late post-engraftment period. In addition, the types and risk factors of infectious complications differ according to the stem cell source, donor type, conditioning intensity, region, prophylaxis strategy, and comorbidities, such as graft-versushost disease and invasive fungal infection. In this review, we summarize infectious complications after HSCT, focusing on the Korean perspectives.
INTRODUCTION
Hematopoietic stem cell transplantation (HSCT), known previously as bone marrow transplantation, is performed to treat two broad categories of diseases. The first category consists of functional failure of bone marrow or marrow-derived cells, including aplastic anemia, myelodysplastic syndrome, immunodeficiency syndromes (severe combined immune deficiency or chronic granulomatous disease), genetic diseases (mucopolysaccharidosis, glycogen storage diseases, etc.), or hemoglobinopathies (thalassemia, sickle cell anemia, etc.). In such cases, HSCT is applied to replace the defective, non-functional bone marrow tissues. Diseases in the second category—which includes hematologic malignancies such as acute or chronic leukemia, multiple myeloma, lymphomas, and myeloproliferative neoplasms—are more common indications for HSCT, the performance of which aims (1) to restore the myelosuppressive or myeloablative effect of cytotoxic treatment (such as intensive chemotherapy and/or total body irradiation [TBI]) to eliminate malignant cells, and (2) to induce a graft-versus-leukemic effect by providing anti-neoplastic immune cells expressing tumor-specific or -associated antigens [1,2].
Until the 1970s, HSCTs were simply classified as autologous or allogeneic according to their donor status, and survival rates were very low. Since the introduction of the human leukocyte antigen (HLA) concept, the incidence of graft failure and/or graft-versus-host disease (GVHD) has decreased. In recent years, allogeneic HSCT has involved various sources of hematopoietic stem cells (bone marrow, peripheral stem cell, cord blood, and mesenchymal cells), donors (sibling, unrelated, haploidentical), and conditioning regimens (standard [myeloablative], reduced intensity). Supportive therapies such as transfusion, colony stimulating factors, and antimicrobial agents have also been developed. Although various early diagnosis and therapeutic techniques have been developed to improve transplant performance, infectious diseases still affect the prognosis of HSCT recipients [2-4].
In Korea, HSCT was first performed in 1983 [5], and the number of transplants has rapidly increased over the past 30 years. In addition to being susceptible to infectious diseases due to neutropenia immediately after HSCT, delayed immune recovery is evident over a long period of time after engraftment, depending on the type of HSCT, immunosuppressants, and acute or chronic GVHD. Because the prevalent infectious diseases vary geographically and over time, state-of-the-art knowledge is needed [6-8]. In this manuscript, infectious complications after HSCT, particularly those common in Korea, are reviewed, as well as the epidemiology, diagnosis, preventive or therapeutic strategies, and novel drugs for improving outcomes of patients.
IMMUNE RECONSTITUTION AFTER HEMATOPOIETIC STEM CELL TRANSPLANTATION
The post-HSCT period is usually divided into the (1) pre-engraftment period (day 0 to days 15–45), (2) immediate post-engraftment period (engraftment to day 100), and (3) late post‑engraftment period (days 100 to 365). In general, the neutrophil count recovers 2 to 3 weeks after HSCT. However, the functional recovery of various cell types is also important. Immunological recovery takes 3 to 6 months for NK cells, 6 to 12 months for B-cells and CD8 T-cells, and 1 to 2 years for CD4 T-cells [9]. The use of T-cell-depleting agents (anti-thymocyte globulin [ATG], alemtuzumab, etc.) to reduce the risk of GVHD and graft failure could improve the outcomes of transplantation but may delay immune recovery [10]. Recipients of T‑cell-depleted transplantation may be vulnerable to viral infections—such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), or adenovirus—and adoptive immunotherapy may be applied [11].
During the pre-engraftment period, the risk of opportunistic infection varies depending on the type of anticancer drugs used, conditioning intensity (myeloablative or reduced intensity), and presence of acute GVHD. Autologous HSCT leads to more rapid recovery of immune function than allogeneic HSCT. In the post-engraftment period, the immune system is reconstituted and recovered in autologous HSCT recipients. However, allogeneic HSCT recipients undergoing long-term immunosuppressive therapy for chronic GVHD remain at risk of infection. Table 1 shows the risk factors for, and infectious diseases commonly encountered after, HSCT.

Common infectious diseases and risk factors according to the various time periods after hematopoietic stem cell transplantation
BACTERIA
Epidemiology
As shown in Table 1, the infectious diseases that occur before the engraftment are similar to those that develop during the neutropenic phase after chemotherapy [2,12]. Neutrophils are important in innate immunity against microorganisms. A decrease in the number of neutrophils increases the susceptibility to infection. In addition, patients with neutropenia have a reduced number of leukocytes. Therefore, inflammatory findings, which are common in patients with normal leukocyte counts, are not often seen, with the exception of fever, which is difficult to diagnose, so the appropriate time to start treatment may be missed [13]. The main sources of bacterial infections in the pre-engraftment phase are the normal gastrointestinal flora and indwelling vascular catheters. Gram-negative bacilli (GNB) are common pathogens in the former, while Gram-positive cocci (GPC) are more common in the latter [14]. In general, microbiologically defined infections (MDIs) account for only 30% to 40% of cases of neutropenic fever [15].
The distribution of causative organisms in febrile neutropenic patients varies geographically. In the United States and Europe, GPC accounted for 60% to 70% of MDI until the early 1990s, while the proportion of GNB increased after the mid-2000s. In a multicenter epidemiological survey, the proportions of Gram-positive and -negative cases were 55% (range, 30% to 85%) and 45% (range, 15% to 70%), respectively [16-18]. Enterobacteriaceae was the most common causative organism, accounting for 30% (range, 8% to 56%). The second most common organism was coagulase-negative staphylococci (24% [range, 7% to 51%]). In Korean studies, GNB were also more common until the mid-2000s, which is consistent with other reports from the Asia-Pacific region [19]. Intestinal bacteria such as Escherichia coli, Klebsiella pneumoniaei, and Pseudomonas aeruginosa are frequently reported GNB, and Enterococcus, Streptococcus, and Staphylococcus species are common among GPC [6-8,20-22]. Because the epidemiology of bacterial pathogens and the resistance profiles varies, early empirical antimicrobial agents must be selected according to the distributions and susceptibilities of the frequently detected bacterial taxa in that center [13,15,19].
In the late post-engraftment phase after autologous HSCT, the immune system is reconstituted and restored, which is consistent with the time at which chronic GVHD typically develops after allogeneic HSCT. Therefore, autologous transplant recipients rarely experience opportunistic infections at this time, but allogeneic HSCT recipients (who take chronic immunosuppressive drugs for several months) are at risk of infection. In this period, infectious diseases may continue to develop. In addition, the immune deficiency resulting from chronic GVHD is associated with infections by encapsulated bacteria such as Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis [2,12]. Deficiencies in immunoglobulin G2 (IgG2) and IgG4 have been reported in patients with chronic and severe GVHD, and are associated with severe pneumonia, meningitis, and sepsis due to S. pneumoniae [23]. Early diagnosis and treatment are essential, and vaccination should be emphasized in this high-risk group of patients. Immunization after HSCT will be described in a separate section of this review.
Resistance
Over the past decade, the frequency of detection of resistant bacteria has increased worldwide, including in patients with neutropenic fever and hematologic malignancies [24]. Among extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae, K. pneumoniae comprises around two-thirds, and E. coli about one-thirds (range, 11% to 69%), depending on the region [18,25-27]. In the late 2000s, 26% of E. coli and K. pneumoniae bacteremia isolates from neutropenic patients were ESBL-producing strains in a single-center study [26]. The incidence of infections caused by carbapenem-resistant Enterobacteriaceae (CRE), along with ESBL-producing Enterobacteriaceae, is increasing in hematologic malignancy patients [18,28-31]. K. pneumoniae, E. coli, P. aeruginosa, and Acinetobacter baumannii are clinically important strains associated with the acquisition of carbapenem resistance. A study of prior colonization by CRE as a risk factor for CRE bloodstream infections found that 45% of neutropenic patients with carbapenem-resistant K. pneumoniae rectal colonization experienced bloodstream infection by an identical strain [30]. There is no multicenter study of CRE in HSCT recipients. However, a single-center, retrospective cohort study performed in Korea reported that the incidence of carbapenem‑resistant A. baumannii bacteremia was 0.53 cases per 10,000 patient-days; post-engraftment infection by this organism can be fatal [31].
The rate of methicillin-resistant coagulase-negative staphylococci is reportedly higher than 50% in most centers, while the incidence rate of Staphylococcus aureus in HSCT patients is low with a high methicillin-resistance rate of median 56% (range, 18% to 100%) [18]. In the late 2000s, coagulase-negative staphylococci were reported to have a methicillin resistance rate of more than 90% and S. aureus a rate of more than 60% [22]. The incidence of Enterococcus bloodstream infections was 1.76 cases per 1,000 patient-days, with vancomycin-resistant Enterococcus (VRE) accounting for 20.6% [22,32]. Most VRE bloodstream infections are caused by E. faecium and are associated with long-term hospitalization and an underlying medical condition, but the resistance itself has not been associated with mortality [32].
The epidemiology of drug-resistant pathogens is important for establishing strategies for prophylaxis and initial empirical antimicrobial therapy. Because domestic data are limited to single-institution and retrospective studies, multicenter nationwide studies of the epidemiology of bacterial infections in Korea are required.
Prophylaxis strategies
Prophylaxis plays an important role during the pre-engraftment phase (Table 2). Oral fluoroquinolone prophylaxis can reduce febrile neutropenic episodes and related mortality in allogeneic HSCT recipients [33,34]. According to the guidelines from the Infectious Diseases Society of America and Infectious Diseases Working Party of the German Society of Hematology and Oncology, ciprofloxacin and levofloxacin are equally effective, but levofloxacin may be more effective in terms of broader coverage of viridans streptococci when oral mucositis is present [15,35]. Although fluoroquinolone prophylaxis has led to an increase in the frequency of drug-resistant (including quinolones) pathogens, the benefit reportedly outweighs the risk [33]. Fluoroquinolone prophylaxis decreased the rate of identification of P. aeruginosa [22]. However, fluoroquinolone prophylaxis is reportedly correlated with resistance development and Clostridium difficile-associated diarrhea [36-41]. Moreover, fluoroquinolone prophylaxis may reduce the rate of bloodstream infections, but not the overall mortality rate [42]. Therefore, the benefit of routine fluoroquinolone prophylaxis should be weighed against its toxicity and effect on the local epidemiology.

Overall infection risk and needs for antimicrobial prophylaxis after HSCT
Empirical therapy: escalation vs. de-escalation therapy
In neutropenic patients during the pre-engraftment phase, infections can progress rapidly. In addition, as it is difficult to distinguish between bacterial infection and non-infectious fever in the early stages, empirical antimicrobial therapy is recommended immediately in all patients with febrile neutropenic episodes. Except for the presence of definite infection foci, the broad-spectrum β-lactam antibiotics ceftazidime, cefepime, piperacillin/tazobactam, and carbapenem are recommended as they have activity against Gram-positive and -negative bacteria, including P. aeruginosa [13,15,43,44]. Step-down therapy can be considered after an initial regimen comprising two or more antimicrobial agents, depending on the local epidemiology and resistance profiles.
The de-escalation approach is generally recommended when high incidence of resistant pathogens in neutropenic fever, or high colonization rates of multiple drug resistance bacteria, or if the patient has a history of infection with drug-resistant pathogens [13,24,44]. However, the data on cut-offs for resistance are insufficient to formulate such a strategy. In addition, no randomized controlled study has compared the therapeutic efficacy of the escalation and de-escalation approaches in HSCT recipients. Selection of the appropriate strategy should take into account the patient’s condition and the prevalence of resistant organisms in the center.
FUNGI
Aspergillus species are the most common causes of invasive fungal infections (IFIs) in patients with hematologic diseases, followed by Candida spp. and rare fungi [45]. The causative fungi are not different after HSCT [46,47]. The characteristics of, and risk factors for, fungal infections differ according to the phase after transplantation. During the pre-engraftment period, neutropenia and mucosal damage are risk factors for invasive candidiasis [4]. Antifungal prophylaxis regimens used during and/or after HSCT are listed in Table 3 [13,48]. Recent advances in the use of prophylactic antifungal agents during HSCT have led to changes in the epidemiology of candidiasis [49]. The Korean epidemiologic data show that the incidence of Candida albicans infection is markedly reduced by antifungal prophylaxis [22]. Nonetheless, the incidence of C. albicans has been decreased by the increasing prophylactic use of fluconazole or echinocandins, and attention should be paid to the selection of antifungal agents for empirical or targeted therapy for invasive candidiasis [50,51]. Post-engraftment, the risk of candida infection decreases with the recovery of neutropenia and mucosal skin loss, but the risk of aspergillosis remains [52].
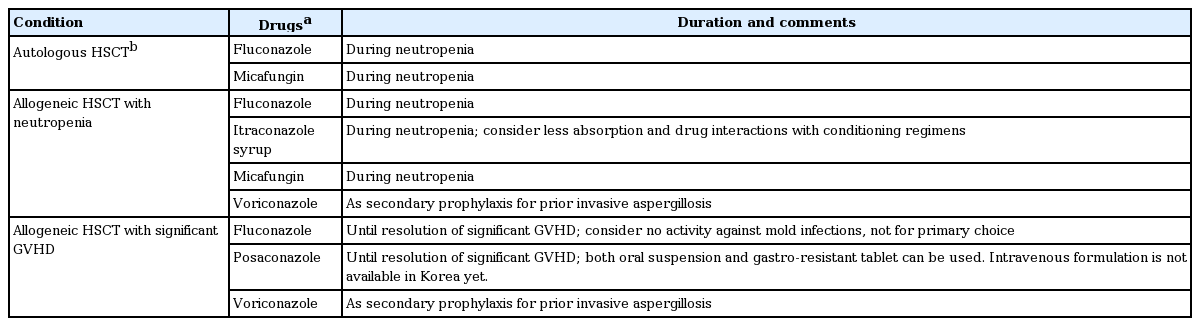
Suggested anti-fungal prophylaxis after HSCT
Invasive aspergillosis (IA) that develops during the neutropenic period typically involves angioinvasion due to host immunosuppression. In contrast, IA that occurs in non‑neutropenic patients, usually due to long-term use of steroids or immunosuppressive agents for GVHD, is more likely to be caused by local inflammation (i.e., airway invasiveness) than by vascular invasion [53]. In the SEIFEM B-2004 study, the most frequent etiological agents of IFI were Aspergillus (70.1%) and Candida (24.8%) species. The IFI-attributable mortality rate was 65.3% (72.4% for allogeneic HSCT recipients and 34.7% for autologous HSCT recipients) [46]. In a prospective observational study performed in the Asia-Pacific region, IA accounted for 65.9% of IFI, followed by 26.7% of invasive candidiasis. Interestingly, Aspergillus spp. was not the major fungal pathogen in centers in Thailand and Vietnam, probably because galactomannan testing was unavailable. The heterogeneity of diagnostic, prophylactic, and therapeutic approaches for IFI necessitates local epidemiological data [54].
The incidence of IFI is higher in allogeneic than in autologous HSCT, and the established risk factors include chronic GVHD and steroid use [3]. According to a recent multicenter study performed in Korea (RISK study), the cumulative incidence of IFI after transplantation is 15.36% per year. Notably, the risk factors were different according to the time after transplantation. Prolonged neutropenia (hazard ratio [HR], 12.61; 95% confidence interval [CI], 4.30 to 36.97, in the early phase), CMV reactivation (HR, 21.44; 95% CI, 2.64 to 173.90, in the late phase), secondary neutropenia (HR, 5.29; 95% CI, 1.14 to 24.49, in the very late phase), and use of secondary immunosuppressive agents due to refractory GVHD (i.e., tumor necrosis factor-α inhibitors) (HR, 37.93; 95% CI, 3.79 to 379.45, in the very late phase) are risk factors for IFIs after HSCT [47]. Therefore, active anti-mold prophylaxis should be considered in patients with those risk factors. Currently, voriconazole is available in Korea for secondary prophylaxis of IA after HSCT, and posaconazole for patients on significant immunosuppressive agents for GVHD.
The diagnosis of IFI after HSCT is not markedly different from that before engraftment. However, the computed tomography (CT) findings of invasive pulmonary aspergillosis in patients with hematologic diseases may differ depending on the time of neutropenia and the time of neutrophil recovery after HSCT, suggesting that the diagnostic criteria may need to be redefined [55]. To diagnose IFIs, radiologic examinations such as chest X-ray and CT, and microbiological studies including fungal culture, and galactomannan and β-D-glucan assays should be performed periodically. For accurate diagnosis, bronchoscopy and bronchoalveolar lavage (BAL) fluid examination, lung biopsy, and culture are required.
Voriconazole is the drug of choice for treating IA. Voriconazole therapeutic drug monitoring can be used in many centers, and the target trough level is 1 to 5.5 mg/L. The CYP2C19 polymorphism affects the pharmacokinetics of voriconazole. Among Korean hematologic patients, 28% are extensive metabolizers (EMs), 48% are heterozygous EMs, and 14% are poor metabolizers. While subtherapeutic initial trough levels were common in EMs, there was no significant relationship between CYP2C19 genotype and the clinical outcomes of IA or the toxicity of voriconazole [56]. Isavuconazole is non-inferior to voriconazole for the primary treatment of suspected invasive mold disease, with fewer drug-related adverse events [57,58]. Salvage therapy includes liposomal amphotericin B, caspofungin, posaconazole, and itraconazole, or a combination of them [58]. Treatment duration is at least 6 to 12 weeks, which should be individualized according to the changes in radiologic, microbiologic, and immunologic parameters.
IFIs can occur during treatment with anti-mold agents. Patients with GVHD under long-term immunosuppressive therapy should be on antifungal agents to prevent IFIs (Table 3). In particular, there is a risk of IFIs other than aspergillosis, such as mucormycosis or other rare molds [48,59,60]. A retrospective single-center study reported that the prevalence and incidence of voriconazole-breakthrough IFIs were 2.25% and 0.22 cases per year, respectively. The overall mortality rate was 44.4% [61]. The possible causes of breakthrough IFI during voriconazole treatment were persistent immunodeficiency, neutropenia, low voriconazole concentration, or poor vascular supply (i.e., abscess or necrotic tissue) [61]. In voriconazole-refractory IA, clinicians should consider the following: misdiagnosis or coinfection with another mold, inadequate blood voriconazole concentration, inadequate tissue drug concentration, immune reconstitution inflammatory syndrome, or infection with voriconazole-resistant Aspergillus [62]. Proven/probable IA patients reportedly have a low culture-positive rate (17.7%) [63]. To determine the azole-resistance rate of Aspergillus clinical isolates in Korea, culture-positive cases should undergo susceptibility testing.
PNEUMOCYSTIS JIROVECII PNEUMONIA
The major symptoms of Pneumocystis jirovecii pneumonia (PCP) are fever, difficulty breathing, and dry cough rather than purulent sputum. Typical radiologic findings are bilateral diffuse infiltrates originating from the periphery of both lungs, but may be normal at the beginning. PCP can present as segmental consolidation in the upper lobe, or subtle ground glass opacities and pneumothorax. It usually occurs within 6 months after transplantation but also after 6 months of immunosuppressive therapy for chronic GVHD [3,4]. The known risk factors for PCP in hematologic patients are acute lymphoblastic leukemia, allogeneic HSCT recipients, alemtuzumab, fludarabine, cyclophosphamide, rituximab, and steroid use (> 20 mg/day prednisone for 4 weeks) [64].
If PCP is clinically suspected, CT, bronchoscopy, and BAL should be performed as early as possible. In a retrospective study of bronchial washings or BAL fluid from non-human immunodeficiency virus (HIV)-infected patients suspected of respiratory infection, among the 169 polymerase chain reaction (PCR)-positive patients, 63.3% were classified as PCP and 46.7% as non-PCP. The majority of patients (84%) in the non-PCP group recovered without treatment for PCP [65]. In two recent meta-analyses, the sensitivity and specificity of quantitative PCR (qPCR) assays were superior to those of non-qPCR assays [66,67]. The serum β-D-glucan test result has a high negative-predictive value but is not useful for the follow-up of PCP [68]. The prognosis is worse in the presence of concomitant infectious diseases, particularly CMV, Aspergillus, and so on. Steroids can be used as an adjunctive therapy in patients with hypoxia (PaO2 ≤ 70 mmHg or PAO2-PaO2 ≥ 35 mmHg).
The incidence of PCP has been significantly reduced by trimethoprim/sulfamethoxazole prophylaxis in highrisk patients. High-risk patients who do not take prophylaxis or who show poor compliance with the drug regimen are the main populations at risk of PCP. In a systematic review and meta-analysis of non-HIV immunocompromised hosts (patients with acute leukemia and recipients of HSCT and solid organ transplant), the incidence of PCP was reduced by 91% (relative risk [RR], 0.09) in trimethoprim/sulfamethoxazole prophylaxis group compared with placebo [69,70]. In addition, PCP-related mortality was significantly reduced by 83% (RR, 0.17). However, trimethoprim/sulfamethoxazole prophylaxis did not markedly reduce all-cause mortality in a hematology population [70]. Mortality rates remain very high in hematology patients (30% to 59%), particularly in HSCT recipients (48% to 70%), compared with 17% to 30% in patients with HIV infection [71]. Nevertheless, given the more severe course of PCP and the higher PCP-related mortality rates, trimethoprim/sulfamethoxazole prophylaxis can likely save lives in other immunocompromised groups as well. PCP should be prevented for at least 6 months or until the immunosuppressant is discontinued in allogeneic HSCT recipients [13,48,71]. Although the optimum duration of PCP prophylaxis is controversial, it is suggested to be continued for a period of time after the immunosuppressant is discontinued. In the setting of corticosteroid-containing regimens, prophylaxis should be continued while steroids are being weaned and/or for 6 weeks after their cessation [72]. With some chemotherapy regimens (i.e., alemtuzumab) consideration should be given to extended PCP prophylaxis for up to 12 months because of the high rate of late-onset PCP [73]. Table 4 summarizes the methods of preventing PCP after HSCT.
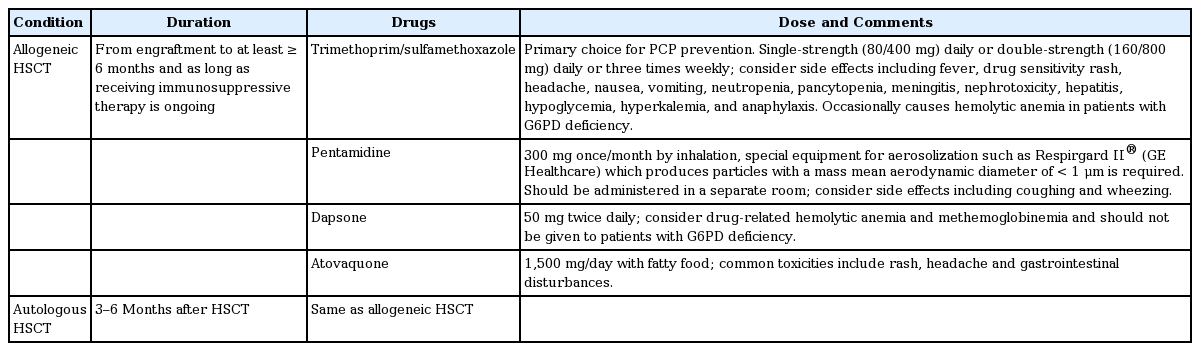
PCP prophylaxis after HSCT
CYTOMEGALOVIRUS
CMV infection can occur after HSCT and can be fatal. CMV can be reactivated or the patient can be reinfected after transplantation. The spectrum of CMV infection is extensive, from CMV reactivation without organ involvement (presenting mainly as asymptomatic antigenemia or DNAemia) to CMV disease such as esophagitis, gastritis, colitis, hepatitis, pneumonia, retinitis, and encephalitis. In addition to direct organ involvement, CMV reactivation can exert indirect effects—such as graft failure or immunosuppression—that may result in the development of concurrent bacterial and/or fungal infections [74]. CMV disease, especially CMV pneumonia or encephalitis, can be fatal despite aggressive anti-CMV therapy [75,76].
A 10-year retrospective single-center study performed in Korea reported that CMV disease occurred in 2.9% of allogeneic HSCT recipients and 0.5% of autologous HSCT recipients. Pneumonia (38.6%), retinitis (36.4%), and enteritis (15.9%) frequently developed in patients with CMV infection. The average time of onset of CMV disease was 90 days after transplantation, ranging from 12 to 936 days [77]. Another study in the same institution reported that CMV DNAemia developed in about 51% of HSCT recipients [78]. CMV reactivation can be associated with a higher non-relapse mortality rate (RR, 1.61 to 1.95) [79]. In another report, 70% of HSCT recipients had experienced any level of CMV antigenemia, of whom 41% had received ganciclovir therapy for significant CMV reactivation and 4% had CMV diseases. However, this incidence could be underestimated because disease prevalence was not evaluated in all patients undergoing pre-emptive therapy [80].
Because the diagnosis of CMV disease is based on the pathology findings, the diagnosis might be limited depending on the patient’s condition. Particularly for the diagnosis of CMV pneumonia, obtaining lung tissue from critically ill patients by transbronchial lung biopsy is problematic. One study aimed to measure the CMV level in bronchial washing fluid and suggest a cut-off for pneumonia diagnosis. CMV DNAemia of > 18,900 copies/mL (137,970 IU/mL) may be associated with CMV pneumonia in post-HSCT patients, as determined by the receiver operating characteristics curve [81]. However, further data are needed because it is difficult to distinguish whether the viral DNA in alveolar hemorrhage reflects viremia rather than lung tissue involvement, or whether CMV is detected as a bystander in bronchial washing fluid.
Management of CMV is categorized into prevention, pre-emptive treatment, and definitive treatment. Pre-emptive therapy is anti-CMV treatment even in the absence of clinical symptoms in cases with CMV infection (reinfection or reactivation). Most transplantation centers introduce pre-emptive therapy rather than routine universal prevention because of insurance coverage, cost-benefit ratio, and adverse drug reactions. Studies of monitoring strategies and early detection have resulted in the use of CMV pp65 antigenemia testing and real-time qPCR for the surveillance and identification of patients suitable for pre-emptive therapy [82,83]. However, in HSCT, the relationship between CMV viral load and CMV disease is different from that in solid organ transplantation. CMV gastrointestinal disease can develop without preceding CMV antigenemia or DNAemia, while > 75,000 copies/mL of CMV DNAemia is reportedly associated with an increased risk of CMV retinitis after HSCT [78,84]. However, the correlations were moderate, and antigenemia or DNAemia does not necessarily precede or accompany CMV disease. Therefore, it is important to identify the at-risk groups and clinical features of the various CMV diseases to facilitate early diagnosis and treatment.
A recent prospective double-blind trial evaluated letermovir prophylaxis for preventing CMV in HSCT recipients. A total of 565 patients underwent randomization and received letermovir or placebo through week 14 after transplantation. The incidence of clinically significant CMV infection was lower in the letermovir group than in the placebo group by week 24 after transplantation (122 of 325 patients [37.5%] vs. 103 of 170 patients [60.6%], p < 0.001). Letermovir prophylaxis resulted in a significantly lower risk of clinically significant CMV infection than placebo. Adverse events with letermovir were mainly of low grade [85]. As a result, the U.S. Food and Drug Administration has approved letermovir for the prevention of CMV infection and diseases in adult CMV-seropositive patients undergoing allogeneic HSCT.
In Korea, which has a high CMV-seropositive rate (95% to 100%) in adults, most donors and recipients are seropositive (D+/R+) and the frequency of CMV infection after transplantation is up to 50% [76]. In addition, owing to active CMV monitoring and pre-emptive therapy, there has been a decrease in the incidence of CMV infection immediately after transplantation. However, late CMV infection (> 3 months after transplantation) is increasing, especially if CMV-specific T-cell function has not been restored due to chronic GVHD. Therefore, immune monitoring is required in patients with high-risk of CMV disease. CMV-specific immune recovery has been studied in kidney transplantation (KT) and HSCT recipients [86-91]. CMV-specific cytotoxic T lymphocytes (CTLs) play an important role in the reconstitution of CMV-specific immunity in immunocompromised patients. The methods used to evaluate CMV-specific immunity include tetramer assay, intracellular cytokine analysis by flow cytometry, measurement of interferon-γ secretion by CMV-specific CD8 T-cells, and enzyme‑linked immunospot assay using IE-1 and pp65 peptide pools [92]. Quantification of CMV‑specific T-cell immunity after HSCT facilitates the identification of patients at risk of CMV‑related complications [87,88].
Currently available anti-CMV agents in Korea are ganciclovir, valganciclovir, foscarnet, and cidofovir. Since February 2018, the use of valganciclovir in HSCT patients with CMV infection has increased due to changes in the reimbursement rules. Regarding adverse events, attention should be paid to bone marrow suppression, and in patients with renal insufficiency. GVHD or the use of monoclonal antibodies (i.e., alemtuzumab) can increase the incidence of CMV infections. Such infections may not be distinguishable from GVHD, or may coexist with GVHD, and may not respond to antiviral drugs if diagnosis is delayed [93]. If there is persistent infection despite anti-CMV therapy, CMV refractoriness can be considered if a > 1 log10 decrease in CMV DNA level in blood or plasma is not achieved after ≥ 2 weeks of treatment. In CMV-refractory cases, resistance should also be suspected. Although CMV resistance remains uncommon in HSCT recipients from HLA-matched donors (0% to 7.9%), in high-risk patients, the incidence of CMV resistance is up to 14.5% [94-96]. The gold standard of CMV resistance testing is an increase in the IC50 (half maximal inhibitory concentration) value by plaque reduction assay. However, CMV resistance can be defined when one or more genetic mutations associated with ganciclovir, valganciclovir, foscarnet, and cidofocir are identified with clinical refractoriness. Therefore, in recent years, mutations in UL97 and UL54 can be tested. Predisposing factors for CMV resistance include prolonged use of anti-CMV agents, recurrent CMV infections, inadequate antiviral absorption, subtherapeutic antiviral level, haploidentical transplantation, or T-cell depletion [97]. If resistance is confirmed, a drug to which resistance has not been identified, or combination of drugs, may be used, but treatment is currently limited. Clinician’s experience is important in such complicated cases.
HERPES ZOSTER
Herpes zoster is caused by the reactivation of virus latent in the posterior ganglia after primary infection with varicella zoster virus (VZV). In Korea, VZV is transmitted by natural infection in most cases and its seroprevalence in adults is > 90%; previous VZV infection is a prerequisite for herpes zoster [98-100]. The clinical features of herpes zoster include abnormal sensation or pain through skin segments 2 to 3 days before skin lesions develop, erythematous spots with irritation, and rapid formation of blisters. The blisters burst and form ulcers, which scar and become dry. Pain and postherpetic neuralgia are major problems. In most cases, one or two dermatomes are involved unilaterally, but dissemination to several other dermatomes or systemically can occur and is associated with visceral or central nervous system involvement [101]. Treatment consists of antiviral administration and adjuvant therapy to reduce the acute pain and postherpetic neuralgia. It is not clear whether antiviral treatment is effective 72 hours after the onset of rash. However, immunocompromised patients with persistent or new vesicular rash, eye involvement, and/or neurologic complications should receive antiviral treatment even > 72 hours after the onset [1].
The risk of varicella is highest in the first 24 months after HCST, or during immunosuppressive therapy for GVHD [101]. In a Korean transplantation center, 34.3% of patients who underwent allogeneic HSCT in 2004 to 2005 experienced herpes zoster. The cumulative incidence was 22% at 1 year post-transplant, and 30.8%, 38.6%, and 41.2% at 2, 3, and 4 years (Fig. 1). Chest dermatomes were most commonly involved (43.8%), and 11.4% of cases were disseminated. There was no herpes-zoster-related mortality.

Cumulative incidence of herpes zoster in allogeneic hematopoietic stem cell transplantation with myeloablative (MAC) or reduced intensity conditioning (RIC) regimens without long-term prophylaxis (log-rank test, p = 0.492).
A live attenuated zoster vaccine is available but cannot be used for HSCT recipients. Acyclovir prophylaxis can reduce the incidence of herpes simplex virus (HSV) and VZV infections not only before engraftment but also in the long term until the immunosuppressant is stopped [102]. It is recommended that acyclovir be maintained for at least 1 year after allogeneic HSCT and for 6 to 12 months after autologous HSCT [3,48]. Antiviral prophylaxis can prevent herpes zoster after allogeneic or autologous HSCT. The pooled RR was 0.29 (range, 0.24 to 0.36) in allogeneic HSCT recipients, and 0.36 (range, 0.25 to 0.54) in autologous HSCT recipients [103].
EPSTEIN-BARR VIRUS AND OTHER HERPESVIRUS INFECTIONS
EBV reactivation can occur 3 to 6 months after transplantation, typically in patients with chronic GVHD. However, progression to disease is relatively rare. Fever and neutropenia may occur as a result of EBV symptoms, which are similar to those of infectious mononucleosis. Most EBV reactivations are subclinical and require no therapy [101]. In addition, aplastic anemia, oral hairy leukoplakia, and post-transplant lymphoproliferative disease (PTLD) can occur. PTLD occurs less frequently after HSCT than after transplantation of other solid organs. EBV-related PTLD occurs in cases of unrelated donor transplantation, T-cell-depleted transplantation, GVHD, and use of an anti-lymphocyte antibody to prevent GVHD [104]. The diagnosis of EBV-associated PTLD can be established by tissue biopsy for histopathology and detection of EBV [105]. Among other herpesviruses, human herpes virus 6 (HHV6) may be responsible for meningitis and hemorrhagic cystitis [106].
HEMORRHAGIC CYSTITIS
Hemorrhagic cystitis in HSCT recipients can be classified according to the onset time (before vs. after engraftment), and cases occurring within 7 days after transplantation are usually non‑infectious. Non-infectious causes include radiotherapy and chemotherapy (e.g., cyclophosphamide, ifosfamide, busulfan, and etoposide), while post-engraftment hemorrhagic cystitis can be caused by virus. Viral reactivation can be accompanied by bladder urothelial damage due to the conditioning regimen. Viral hemorrhagic cystitis can be caused by polyomaviruses (BK and JC viruses), adenovirus, CMV, HSV, and HHV6 [107,108].
Of these viruses, BK virus (BKV) is the most common; 80% to 90% of adults are BKV seropositive [109-111]. BKV-associated hemorrhagic cystitis is predominant in allogeneic HSCT patients, and BKV-associated nephropathy in KT patients. Why BKV reactivation manifests as these two major forms in KT and HSCT patients is unclear [112]. The diagnostic criteria defined by the European Conference on Infections in Leukemia (ECIL)-6 comprise the following triad: viral replication (urine BKV > 107 copies/mL), symptoms of cystitis, and hematuria of grade II or higher. A blood BKV DNA level of > 103–4 copies/mL is reportedly associated with significant viruria. However, a negative plasma viral load does not rule out BKV-associated hemorrhagic cystitis [113-115].
The risk factors for hemorrhagic cystitis are myeloablative conditioning regimen, ATG, and GVHD. The incidence is higher in allogeneic than in autologous HSCT patients, and in adults than in pediatric cases. Treatment for virus-associated hemorrhagic cystitis should be initiated by minimizing immunosuppressants. Cidofovir might be effective for adenovirus, CMV, and BKV, and can be considered if there is ≥ grade III hematuria (gross hematuria with blood clots) and no response to conservative treatment [113].
In eight retrospective and two prospective studies, the use of cidofovir with or without probenecid (either 3 to 5 mg/kg with probenecid or 0.5 to 1.5 mg/kg without probenecid) was investigated. A reduction in the urine and blood BKV load reportedly occurs in 38% to 62% and 67% to 84% of patients, respectively [116-122]. A retrospective study did not recommend levofloxacin because of insufficient data and the low level of evidence [113]. In addition, retrospective data are available for vidarabine, leflunomide, hyaluronic acid, and mesenchymal cells, but these are not recommended by the guidelines [113]. Brincidofovir markedly reduces BKV replication in vitro [123]. In cases of persistent hematuria, renal function can be impaired by blood clots and related post-renal obstructions. Conservative care such as intravenous hydration and removal of obstructions is also important.
COMMON RESPIRATORY VIRUSES
Epidemiology and risk factors for progression to lower respiratory infections
Common respiratory virus (CRV) infections are frequent causes of upper respiratory tract infection (URTI) and lower respiratory tract infection (LRTI) in HSCT patients. CRV infections can also occur during neutropenia and show significant morbidity and mortality. The epidemiology of CRV infection in HSCT patients is likely to reflect that in the community, with seasonal variations. Influenza and respiratory syncytial virus (RSV) infections usually occur in winter, parainfluenza virus (PIV) infections in summer, and rhinovirus throughout the year. In HSCT recipients, CRV infection is not limited to URTIs and is more likely to progress to LRTIs [124,125]. Respiratory virus multiplex PCR enables rapid diagnosis of CRV infections in clinical practice [126].
There are little data on CRV infection in HSCT patients in Korea. In a 4-year retrospective study in the authors’ HSCT center from 2007 to 2011, 67 of 1,038 HSCT patients (6.5%) had 71 cases of CRV-LRTI. RSV (43.6%) was the most common pathogen of CRV-LRTI, followed by PIV (26.8%), influenza virus (19.7%), and rhinovirus (9.9%) [127]. The overall mortality rate at day 30 after CRV-LRTI was 32.8%, and high-dose steroid usage (> 1 mg/kg/day), severe immunodeficiency, and lymphopenia (absolute lymphocyte count < 200 cells/mm3) were significantly associated with mortality [127]. These findings are similar to the prevalence, mortality rate, and mortality-related risk factors of CRV-LRTI in Europe and the United States.
The average mortality rate of RSV-LRTI is 32% (range, 0% to 70%) in international studies, and the major risk factors for progression to LRTI are lymphopenia, old age, mismatched/unrelated donor, and neutropenia [128,129]. In cases of PIV-LRTI, the overall mortality rate is 10% to 30%, and high-level corticosteroid exposure, neutropenia, lymphopenia, and early onset after HSCT are the major risk factors for LRTI [129-131]. The rate of progression of influenza to LRTI is 25% to 28% and the overall mortality rate is 25% to 58% [132-134]. The risk factors for progression to influenza-LRTI are early onset after HSCT, lymphopenia, old age, neutropenia, and delayed antiviral administration [132-134]. In cases of rhinovirus, most infections are asymptomatic, less than 10% of patients progress to definite pneumonia, and the mortality rate is < 10% [135,136]. In addition, human metapneumovirus, adenovirus, coronavirus, and bocavirus can cause URTIs and LRTIs in HSCT patients [129,137].
Treatment
Management of CRV infection in HSCT patients can be classified into four categories: prevention (infection control), selective therapy for URTI, antiviral therapy, and supportive care. Currently, a commercialized vaccine is available only for influenza, and so preventive strategies to stop the spread of CRV infections—such as hand hygiene, wearing gloves and mask, and isolation of symptomatic patients—have been emphasized [129,134,137,138]. It is recommended that healthcare workers and family members of patients be vaccinated against influenza, as well as undergo post-exposure prophylaxis (in the Korea influenza guidelines, oseltamivir can be administered prophylactically for 10 days if an immunocompromised patient comes into contact with a patient with confirmed influenza or influenza-like illness) [134,137-139].
Because some CRV-URTIs do not progress to LRTI and therapeutic agents are limited, it is important to identify CRV‑URTI patients at increased risk of LRTI [129,137,140]. Treatment of RSV-URTI with aerosolized ribavirin significantly reduces the frequency of progression to LRTI (25% vs. 47%) [128]. In addition, the following immunodeficiency scoring index has been proposed: neutropenia (< 500 cells/mm3, 3 points), lymphopenia (< 200 cells/mm3, 3 points), age ≥ 40 years (2 points), myeloablative conditioning (1 point), GVHD (acute/chronic, 1 point), corticosteroids (1 point), and pre-engraftment or within 30 days of transplant (1 point). A score of > 7 is associated with a significantly increased risk of progression to LRTI, suggesting the necessity of criteria for identifying high-risk groups [141]. Palivizumab, a humanized anti-RSV monoclonal antibody, does not reduce the progression to LRTI in HSCT patients and is currently recommended only for prophylactic usage in high-risk children [142]. The risk factors for progression to LRTI have been discussed for PIV, human metapneumovirus, and rhinovirus URTIs, but treatment is not recommended because of a lack of evidence [129,137,140].
Oseltamivir, zanamivir, and peramivir can be used to treat influenza URTIs and LRTIs and should be administered within 48 hours of symptom onset [134,139]. Delayed administration is also beneficial, so these agents should be administered even after 48 hours [134,139]. Although there is no definitive evidence, most guidelines recommend that antiviral treatment be extended beyond the usual duration in cases of severe influenza LRTI [134,139,143]. Aerosolized ribavirin and intravenous immunoglobulin (IVIG) combination treatment significantly reduces RSV-LRTI-related mortality and is thus recommended by most guidelines [129,137,140,141]. Although evidence is lacking, if aerosolized ribavirin cannot be used, oral ribavirin or intravenous ribavirin may be considered [129,137,140,141]. A recent double-blind, placebo‑controlled study of GS-5806 (presatovir), an oral RSV-entry inhibitor, involved 54 RSV-infected healthy adult volunteers who received RSV challenge intranasally. GS-5806 reduced the viral load and the severity of clinical disease [143]. Proven PIV-LRTI should be treated with aerosolized ribavirin and IVIG in combination [129,137,140]. Unfortunately, aerosolized ribavirin is costly and available only through the Korea Orphan & Essential Drug Center in Korea, so its use is limited. No antiviral is available to treat CRV, rhinovirus, human metapneumovirus, and bocavirus LRTIs, so supportive care is the mainstay.
HSCT patients with CRV infection are often susceptible to other pathogens, and so work-up for co-existent pathogens is required [129,137,140]. S. aureus and S. pneumoniae infections are commonly followed by influenza infection. Therefore, patients with influenza LRTIs should be administered antibiotics to which these bacteria are susceptible [144]. As it is not possible to distinguish these bacteria from other co-pathogens (i.e., PCP, CMV, or fungi, etc.) using only imaging tests and clinical symptoms, examinations such as bronchoscopy are required [129,137,140].
TUBERCULOSIS
Tuberculosis (TB) is an important opportunistic infection among HSCT recipients, in whom its incidence is 2- to 40-fold higher than that in the general population; however, this is lower than in solid-organ-transplant patients [145-147]. HSCT recipients are immunosuppressed as a result of their hematologic disease, chemotherapy, radiotherapy, immunosuppressive agents, and GVHD [146]. Several risk factors for the development of TB after HSCT have been reported, including acute myeloid leukemia, chronic myeloid leukemia, myelodysplastic syndrome, busulfan, cyclophosphamide, TBI, corticosteroid therapy, HLA-mismatched transplant, GVHD, or a history of TB infection [148]. Of HSCT patients who develop TB, 80% and 20% had undergone allogeneic and autologous transplant, respectively.
A single-center study performed in Korea reported that 2.5% of allogeneic HSCT recipients were diagnosed with TB after HSCT. The median time to development of TB was 386 days after transplantation (range, 49 to 886). The standardized incidence ratio of TB, compared with that in the general population, was 9.10 (95% CI, 5.59 to 14.79). Extensive chronic GVHD was associated with the development of TB (p = 0.003) (Fig. 2) [146]. Another center in Korea reported that HSCT patients with TBI-based conditioning are likely to have TB disease [149]. Extrapulmonary TB comprises 42% of post-HSCT TB cases, which is higher than that in the general population (15% to 20%) [146].

Cumulative incidence of tuberculosis (TB) in allogeneic hematopoietic stem cell transplantation (HSCT) recipients with and without chronic graft-versus-host disease (cGVHD) (p = 0.003).
Owing to the risk of reactivation or new infection, prophylaxis is recommended for HSCT recipients with a positive TB-specific interferon-γ release assay result [3]. Isoniazid is well tolerated in the post-HSCT period. However, concurrent itraconazole is not recommended due to drug interactions, and the impact of voriconazole or posaconazole is unclear. Isoniazid should be continued for at least 9 months until immunosuppression is reduced. Rifampin may undergo drug-drug interactions with immunosuppressants, which makes this option not practical. To determine the optimum timing and duration of isoniazide prophylaxis in HSCT patients, further studies are needed.
POST-HSCT VACCINATION
Post-HSCT vaccination is recommended regardless of the type of transplantation and source of stem cells. Guidelines recommend that both allogeneic and autologous HSCT recipients should be immunized as scheduled even during immunosuppressive therapy for chronic GVHD [150,151]. However, if > 0.5 mg/kg prednisolone is administered for GVHD, it may be temporarily delayed to increase the immune response until the dosage of immunosuppressants become decreased. The recommended schedules in Korea are presented in Table 5 [152].
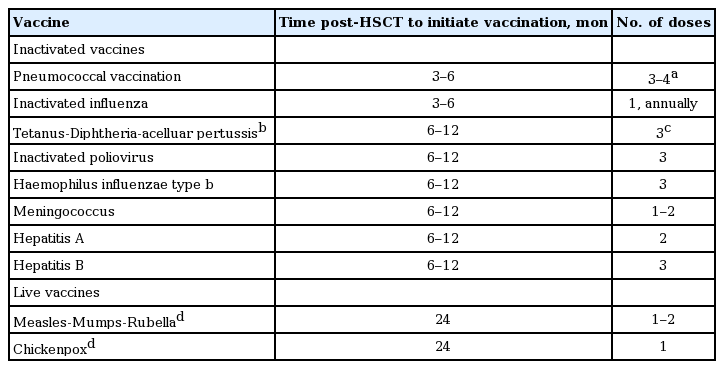
Recommended vaccination for HSCT recipients
CONCLUSIONS
HSCT is performed to treat various hematologic malignancies and other disorders. Although survival rates have improved due to developments in transplantation techniques, infectious complications remain major causes of morbidity and mortality after HSCT. The severity of infectious complications varies according to the phase post-transplant, type of transplantation, GVHD, and the degree of immunosuppression or reconstitution. Further studies are required to improve the treatment of infectious diseases, and the outcomes of HSCT patients are expected to improve in future.
Notes
No potential conflict of interest relevant to this article was reported.