Rapid onset of efficacy predicts response to therapy with certolizumab plus methotrexate in patients with active rheumatoid arthritis
Article information
Abstract
Background/Aims
The objective of this study was to determine the efficacy and safety of add-on therapy with certolizumab pegol (CZP) in active rheumatoid arthritis (RA) patients of a single ethnicity.
Methods
In this 24-week, phase 3, randomized, double-blind, placebo-controlled trial, eligible patients (n = 127) were randomized 2:1 to subcutaneous CZP + methotrexate (MTX; 400 mg at week 0, 2, and 4 followed by 200 mg every 2 weeks) or placebo + MTX.
Results
At week 24, the American College of Rheumatology criteria for 20% (ACR20) response rate was significantly greater with CZP + MTX than with placebo (66.7% vs. 27.5%, p < 0.001). Differences in ACR20 response rates for CZP vs. placebo were significant from week 1 (p < 0.05) and remained significant through week 24. The CZP group reported significant improvement in physical function and disability compared to the placebo group (p < 0.001) at week 24, as assessed by Korean Health Assessment Questionnaire-Disability Index (KHAQ-DI). Post hoc analysis indicated that the proportion of patients who had ACR70 responses, Disease Activity Score 28 (DAS28) low disease activity, and DAS28 remission at week 24 was greater in CZP + MTX-treated patients who achieved a decrease in DAS28 ≥ 1.2 (43.8%) at week 4 than in nonresponders. Among 18 (22.2%) and 14 patients (35.0%) in CZP and placebo groups who had latent tuberculosis (TB), none developed active TB. Most adverse events were mild or moderate.
Conclusions
CZP treatment combined with MTX in active RA patients with moderate to severe disease activity and an inadequate response to MTX resulted in rapid onset of efficacy, which is associated with better clinical outcome at week 24 and has an acceptable safety profile, especially in an intermediate TB-burden population.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disease associated with significant morbidity and mortality. The treatment of RA has shown remarkable advancement since the development of tumor necrosis factor (TNF) inhibitors added an effective armament on the synthetic disease-modifying anti-rheumatic drugs (DMARDs), such as methotrexate (MTX) and hydroxychloroquine [1]. In addition to the new targeted biologics, the refinement of TNF inhibiting agents has been underway, which includes a fully human anti-TNF antibody [2] and a PEGylated anti-TNF Fab antibody [3], certolizumab pegol (CZP).
CZP has shown clinical efficacy in the treatment of RA with moderate to severe disease activity either as a monotherapy or in combination with MTX and other DMARDs [3-5]. The combination of CZP and MTX was superior in the prevention of radiographic progression and in the improvement of physical function compared with MTX monotherapy [3,4]. Improvements in clinical outcomes have been achieved from the earliest visit after beginning of therapy (typically week 1) [3,4].
Previous studies have shown that reducing clinical disease activity as much as possible may result in the maximal reduction of disability [6-8]. In addition, earlier response to treatment is predictive of the long-term outcome [6]. Disease activity at week 12 of treatment with TNF inhibitors has been related to long-term clinical and radiographic outcomes [9]. Furthermore, rapid attainment of clinical response at week 6 was associated with faster, more sustained improvements in patientderived outcomes and with a better chance of achieving remission at week 52 compared to response at week 12 [10].
This study was a phase 3, 24-week, randomized, double-blind, placebo-controlled trial in which patients with moderate to severe RA and inadequate response to MTX could be enrolled. The purpose of this study was to determine the efficacy and safety of CZP in RA patients from a single ethnicity. In addition, this study analyzed whether early response to treatment is associated with better 24-week clinical outcome.
METHODS
Study setting and design
This was a 24-week, phase 3, double-blind, randomized, multicenter, placebo-controlled study conducted from December 2009 through August 2011 at 15 centers in Korea (NCT00993317). The Institutional Review Board (KNUH_09-0078) at each participating center approved the study protocol. All patients provided written informed consent, and the study was conducted in accordance with the principles of the Declaration of Helsinki as well as the relevant regulations and guidelines [11]. Eligible patients were randomized 2:1 to receive either CZP 200 mg subcutaneously every 2 weeks following a loading dose of CZP 400 mg at week 0, 2 and 4, or placebo (0.9% saline), plus MTX. All patients continued MTX treatment at the same dose and route of administration as at entry. Patients who failed to achieve a response according to the American College of Rheumatology criteria for 20% (ACR20) improvement at week 12 (and confirmed at the week 14 visit) were considered treatment failures. After the week 14 dosing, these patients and patients who completed the study at week 24 were offered the choice of entering the open-label extension (OLE) study. If the patients elected to enter the OLE study, they were to complete the assessments at week 24/withdrawal assessments at week 16 of the present study.
Participants
Eligible patients were aged 18 to 75 years with a diagnosis of RA, as defined by ACR 1987 criteria [12], for at least 6 months duration but not longer than 15 years. Patients had to have active disease at screening and at baseline, defined by ≥ 9 tender joint count based on 68 joints, ≥ 9 swollen joint count based on 66 joints and either erythrocyte sedimentation rate (ESR) ≥ 30 mm/hr (Westergren) or C-reactive protein (CRP) ≥ 15 mg/L [13]. MTX had to be stable for at least 8 weeks prior to baseline visit with a minimum dose of 10 mg weekly.
Exclusion criteria included any active infection (including currently active tuberculosis [TB]), positivity for hepatitis B virus surface antigen (HBsAg), hepatitis C antibody (HCV Ab), or human immunodeficiency virus (HIV) serum testing, uncontrolled medical diseases, and rheumatic diseases other than RA. Patients were also excluded if they had received any biological therapy for RA within 24 weeks prior to baseline (12 weeks for etanercept), had a severe hypersensitivity or anaphylactic reaction with a biological agent, or had been resistant to previous anti-TNF therapy. Oral corticosteroids had to be stable for at least 4 weeks and during the trial with maximum dose of 10 mg/day prednisolone equivalent. Doses of nonsteroidal anti-inflammatory drugs were fixed for 2 weeks prior to baseline visit and during the trial.
Patients were screened for TB according to the guidelines using skin test with purified protein derivatives (PPD) and chest X-ray. If the PPD skin test was positive (induration ≥ 5 mm) or the patient had had close contact with patients with active TB, but there was no clinical or radiologic evidence of TB, patients could be enrolled into the trial after adequate treatment for latent TB with isoniazid (up to 300 mg) initiated at least 1 month before randomization.
Efficacy assessments
Efficacy assessments were carried out at baseline and at week 1, 2, 4, 6, 8, 12, 14, 20, and 24/withdrawal. The primary efficacy endpoint was the ACR20 response at week 24. The secondary efficacy variables were ACR20 response at week 12, ACR50/ACR70 response at week 12 and 24, and Korean Health Assessment Questionnaire-Disability Index (KHAQ-DI) at week 24 [14,15]. Other efficacy variables included ACR20/ACR50/ACR70 response rates at other time points, ACR core components, health-related quality of life (HRQoL) as measured by the Short Form-36 (SF-36) health assessment tool [16], Disease Activity Score in 28 joints calculated by using ESR (DAS28 [ESR]), European League Against Rheumatism (EULAR) response criteria, and ESR [17].
Among the secondary efficacy endpoints, patient-reported outcomes (PRO) included pain, patient’s global assessment of disease activity, physical function assessment using the KHAQ-DI, and HRQoL assessed using the SF-36 Health Survey.
Safety evaluations
Safety was assessed based on reported adverse events (AEs) which were assessed at all visits and at 12-week follow-up after the last dose of investigational product. Physical examination, laboratory tests, and chest radiography were monitored at predefined schedule. AEs that required hospitalization, resulted in death, or caused disability were defined as serious AEs (SAEs). For immunologic analyses, auto-antibodies such as anti-double stranded DNA and antinuclear antibodies were measured at baseline and week 24/withdrawal visit and anti-CZP antibodies at baseline, week 1, 2, 4, 6, 8, 12, and 24.
Statistical methods
The sample size was determined using predicted rate of ACR20 response with 2:1 randomization ratio for CZP and placebo groups on the assumption of ACR20 response of 50% in the CZP group and 21% in the placebo group. A sample size of 80 and 40 patients in CZP and placebo groups respectively was estimated to be required to provide 90% power for a two-tailed test with an alpha of 0.05.
Efficacy analyses were performed using the full analysis set (FAS) population (n = 121). For the responder analyses, patients who withdrew for any reason or used rescue medication were considered as nonresponders from that time point onwards (nonresponder imputation). Sensitivity tests were carried out on the data imputed using the last observation carried forward (LOCF) method. All secondary and other endpoints, apart from responder analyses, were imputed using LOCF. Where analysis of covariance was carried out on an efficacy endpoint, baseline value was included in the model as a covariate and treatment group as a factor. Comparisons between the CZP + MTX and placebo + MTX groups were performed using logistic regression with treatment group as a factor. Odds ratios and 95% confidence intervals are presented. p values of < 0.05 were considered to be statistically significant.
Safety and tolerability analyses were performed in the safety population in all randomized patients who received at least 1 dose of study drug. The number and percentages of patients reporting AEs were tabulated and analyzed per the Medical Dictionary for Regulatory Activities coding terms.
RESULTS
A total of 127 patients were randomized into the study. Of these, 121 patients (FAS) received either CZP (n = 81) or placebo (n = 40) plus MTX (Fig. 1). Substantially fewer patients were withdrawn due to lack of efficacy or AEs related to worsening of RA disease in the CZP + MTX group compared to placebo + MTX group (45% vs. 21%). At week 24, 58 patients (71.6%) in the CZP group completed the study compared with 20 patients (50.0%) in the placebo group. The mean age, percentage of female, body mass index, mean duration of RA, and rheumatoid factor positivity were similar between the groups (Table 1).

Disposition of patients. FAS, full analysis set; MTX, methotrexate; CZP, certolizumab pegol. aCZP 200 mg every 2 weeks is preceded by CZP 400 mg at weeks 0, 2, and 4.
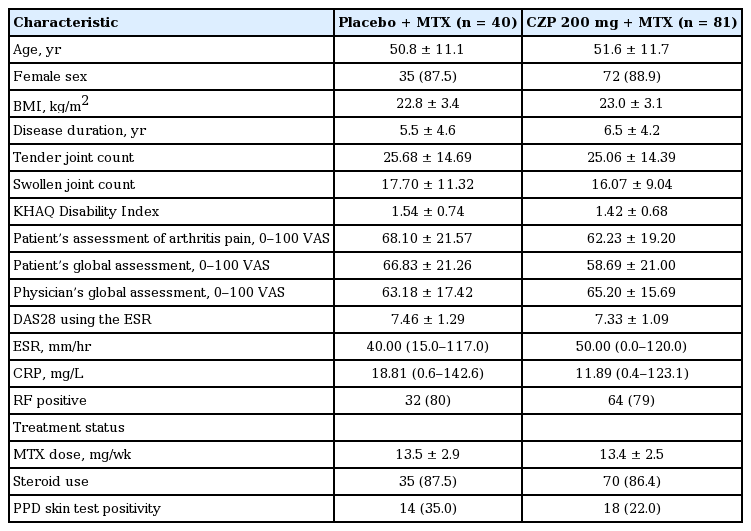
Baseline characteristics of the FAS population
At week 24, the ACR20 response rate (primary end-point) was significantly greater in the CZP + MTX group compared to the placebo + MTX group (66.7% vs. 27.5%, p < 0.001) (Fig. 2A). ACR20 response rate at week 12 was also statistically significant for the CZP + MTX group compared to the placebo + MTX group (64.2% vs. 37.5%, respectively; p = 0.006). Meaningful differences in ACR20 response rates between treatment groups began at week 1 (p = 0.013) and remained through week 24 (p < 0.05 for each comparison). The slope of increase in ACR20 response rate between time points was most steep at week 1, followed by week 2 and week 4 which were then followed by sustained but slower improvements. The ACR50 and ACR70 responses at week 24 were statistically significant when comparing the CZP + MTX group with the placebo + MTX group (43.2% vs. 20.0%, p = 0.014 and 17.3% vs. 2.5%, p = 0.047, respectively) (Fig. 2A). One patient in the placebo + MTX group and 14 patients in the CZP + MTX group achieved an ACR70 response at week 24 (Fig. 2B). A treatment difference in improvement in DAS28 was observed by week 1 for patients receiving CZP + MTX compared to placebo + MTX (mean change from baseline: –0.70 vs. –0.22, respectively; p < 0.001), and was maintained until week 24 (–2.03 vs. –0.94, p < 0.001) (Fig. 2C). For the CZP + MTX group, by week 4, the mean change of DAS28 from baseline approached half of the improvement seen at week 24. EULAR good/moderate response rates at weeks 4 and 24 for CZP + MTX group were 56.5% and 81.3%, respectively; versus 15.4% and 48.7%, respectively, for placebo + MTX group (Fig. 2D).

Clinical efficacy of certolizumab pegol (CZP) + methotrexate (MTX) or placebo (PBO) + MTX in the treatment of patients with active rheumatoid arthritis for 24 weeks. (A) American College of Rheumatology (ACR) 20%, 50%, and 70% response criteria (ACR20, ACR50, and ACR70) response rates at week 24. (B) ACR20, ACR50, and ACR70 response rates over time, nonresponder imputation (NRI). (C) Mean changes from baseline in the Disease Activity Score in 28 joints (DAS28; erythrocyte sedimentation rate [ESR]). (D) European League Against Rheumatism (EULAR) response criteria good/moderate response rates over time, NRI. ap ≤ 0.05, bp ≤ 0.001 (vs. PBO + MTX, NRI), and cp ≤ 0.05, dp ≤ 0.001 (vs. PBO + MTX at each time point, last observation carried forward).
We next questioned whether responses at as early as week 4 would influence week 24 outcomes. At week 4, both ACR20 and EULAR good/moderate response rates were higher than 50% and the mean change of DAS28 from baseline was approximated as almost half of the improvement seen at week 24 for CZP + MTX group. Responders were then defined as patients achieving a decrease in DAS28 (ESR) of ≥ 1.2 from baseline. Post hoc analysis revealed that the proportion of patients who had ACR70 responses, DAS28 low disease activity (LDA; defined as a DAS28 ≤ 3.2), and DAS28 remission (defined as a DAS28 ≤ 2.6) at week 24 was greater in the population of week 4 responders than in nonresponders (Fig. 3). CZP + MTX-treated patients who achieved a DAS28 ≥ 1.2 (43.8%) at week 4 had similar demographics and disease characteristics at baseline (data not shown) compared with nonresponders.

Proportion of American College of Rheumatology (ACR) 70 responders, patients with low disease activity (LDA), and patients in Disease Activity Score in 28 joints (DAS28) remission at week 24 in patients who achieved a ≥ 1.2-point reduction compared with patients who achieved a < 1.2-reduction in DAS28 at week 4.
In addition to clinical outcomes, CZP + MTX provided measurable improvements in PROs, including pain, patient’s global assessment for disease activity, KHAQ-DI, and HRQoL over placebo + MTX. At week 1, CZP + MTX group reported significant improvement in pain, patient’s global assessment, and KHAQ-DI. The mean change from baseline in KHAQ-DI at week 24 was –0.54 with CZP + MTX compared to –0.17 with placebo + MTX (p < 0.001) (Fig. 4). Patients in the CZP + MTX group showed greater improvements from baseline in HRQoL as assessed by Physical Component Summary score (PCS) and Mental Component Summary score (MCS) of SF-36 compared with those in the placebo + MTX group. The differences between treatment groups were small for the PCS compared to the MCS, although all SF-36 domain sub-scores were numerically improved in patients treated with CZP + MTX compared to placebo + MTX. In a post hoc analysis, DAS28 response at week 4 was associated with clinically significant improvements in PROs such as pain and patient’s global assessment at week 24.

Mean change from baseline in Korean Health Assessment Questionnaire-Disability Index (KHAQ-DI) until week 24. PBO, placebo; MTX, methotrexate; CZP, certolizumab pegol. ap ≤ 0.05, bp ≤ 0.01, and cp ≤ 0.001 vs. PBO + MTX at each time point, last observation carried forward.
Of the 127 patients in the safety set (patients who received at least one injection of study drug), a total of 164 treatment emergent adverse events (TEAEs) were reported in 56 patients (65.9%) treated with CZP + MTX, whereas 60 TEAEs were reported in 24 patients (57.1%) treated with placebo + MTX (Table 2). The majority of AEs in both treatment groups were mild to moderate in nature. The most frequently reported adverse events were infections, followed by gastrointestinal disorders and injection site reactions. Infectious AEs were reported at similar rates in the CZP + MTX group (34.1%) and the placebo + MTX group (28.6%). The most frequent infectious AEs were upper respiratory tract infections and urinary tract infections. AEs leading to withdrawal from the study were reported in CZP + MTX patients (n = 4, 4.7%) and placebo + MTX patients (n = 2, 4.8%). A total of 15 SAEs were experienced by eight patients (9.4%) in the CZP + MTX group compared to none in the placebo + MTX group. The most common SAEs were infections occurring in six CZP-treated patients. At baseline, 18 (22.2%) and 14 patients (35.0%) in the CZP + MTX and placebo + MTX groups, respectively, had PPD indurations ≥ 5 mm in diameter (Table 1). All of these patients except for one received prophylactic treatment for TB. One subject in the placebo + MTX group did not receive TB prophylaxis despite PPD positivity and was reported as a protocol deviation. This subject didn’t experience TB during the study. Two patients developed TB after 147 and 167 days of exposure to CZP. Both patients were PPD negative at baseline.

Summary of AEs
There were no cases of death, demyelinating diseases, malignancy, or lupus-like syndrome. The incidence of conversion of antinuclear antibody from negative at baseline to positive at week 24 was higher in the CZP + MTX group (11.1%) than the placebo + MTX group (7.1%). One patient (2.4%) in the placebo group converted anti-double stranded DNA (anti-dsDNA) antibody from negative at baseline to positive at week 24. Anti-CZP antibodies were detected in 1.3% and 3.8% of CZP + MTX group at week 12 and week 24, respectively.
DISCUSSION
This 24-week randomized controlled trial demonstrated that addition of CZP (400 mg at week 0, 2, and 4, followed by 200 mg every other week) to MTX in active RA patients who had a previous inadequate response to MTX alone rapidly improved disease activity. This study was performed in patients of single ethnicity with an intermediate TB burden, and demonstrated a similar efficacy of CZP treatment compared with that of previous studies on a multi-ethnicity background [3,4], and comparable risks for AEs including TB infection.
The primary endpoint, ACR20 response rate at week 24, was 66.7% in the CZP + MTX group, which was slightly higher compared to 58.5% and 57.3% in Rheumatoid Arthritis Prevention of structural Damage 1 (RAPD1) [3] and RAPID2 [4] trials, respectively, but the response rate of placebo group was also higher in this study. The odds of being an ACR20 responder at week 24 was 5.3 times greater for patients in the CZP 200 mg plus MTX group compared to the placebo + MTX group (95% confidence interval, 2.3 to 12.1; p < 0.001). The efficacy of CZP + MTX was also demonstrated for all secondary and other endpoints, showing rapid response as early as week 1 and consistent improvement throughout week 24 compared to placebo + MTX. A rapid change in ACR20 response rate, DAS28 reduction and KHAQ-DI improvement continued until week 4, which was followed by slower but continuous improvements at subsequent visits.
At week 24, a higher percentage of patients in the CZP + MTX group achieved ACR50 and ACR70 response compared to those in the placebo + MTX group. CZP + MTX treated patients reported significant improvement in physical function and disability compared to placebo + MTX patients (p < 0.001), as assessed by the KHAQ-DI. Furthermore, CZP-treated patients showed an improvement from baseline in health-related quality of life, as assessed by SF-36 PCS and MCS.
In a post hoc analysis, patients that had decreased DAS28 ≥ 1.2 from baseline response at week 4 to CZP + MTX showed a better response in clinical outcomes and PROs at week 24, revealing that early clinical response to CZP treatment is associated with good clinical responses in terms of ACR70, LDA, and DAS28 remission after longer term treatment at week 24. Although it was not the focus of the present trial, it was of interest that the earlier responders at week 4 had a better clinical outcome measure. Previous studies showed that the level of disease activity at 3 months was related to the increased possibility of LDA or remission after 1 year of treatment with TNF inhibitors [6]. Furthermore, patients with an earlier clinical response to CZP + MTX at week 6 were more likely to have a better long-term clinical response at week 52 [10]. The response to anti-TNF treatment at week 6 may predict drug discontinuation at 3 months [18]. These results including the present study implicate that the faster response to TNF blockade predicts improved long-term clinical outcome, which may help reduce unnecessary drug exposure by earlier determination of alternative therapy [10].
CZP + MTX in the present trial had a comparable safety profile to other anti-TNFs, with a low incidence of discontinuations due to AEs [3,4,19-21]. The percentage of CZP patients reporting infectious AEs was similar to those reported for other TNF inhibitors in a population of the same ethnicity [22] and in heterogeneous populations of multi-ethnicity [3-5]. There were no deaths or malignancy among patients who participated in this study.
The incidence of serious infections, including TB, in the CZP + MTX group was consistent with those reported with anti-TNF treatments [23,24]. In the present study which was performed in an intermediate TB-burden population (92/100,000 general population) [25], TB occurred in two patients with CZP treatment. Although patients who were considered to have a risk of TB were treated with a prophylactic regimen based on the current clinical procedures using guidelines for latent TB treatment during TNF antagonist use [26,27] TB may still occur in patients with negative PPD skin tests, representing new infections rather than reactivations [28]. The comparable incidence of TB in this study with the trials of TNF inhibitors performed in mixed TB-burden populations indicates the safety of CZP therapy in patients living in countries with an intermediate prevalence of TB, if appropriate prophylactic measures are applied [4,19-21]. Thus, CZP + MTX has an acceptable safety profile for the treatment of patients with RA which was in line with other reports of CZP [3,29] and with reports of other anti-TNFs in the same population [22].
Limitations of this study include the short duration of 24 weeks, small size of study population and lack of radiographic outcome measures. Further long-term data by the OLE to this study and additional registry programs may assist in identification of better predictors of long-term clinical outcome.
In conclusion, combined treatment with CZP in patients with active RA and an inadequate response to MTX resulted in rapid onset of efficacy which is associated with better clinical outcome at week 24 and has an acceptable safety profile, even in an intermediate TB-burden population with adequate TB prophylaxis management. Together with established efficacy, these results augment the available evidence that CZP treatment is safe and effective in the treatment of RA patients with moderate to severe disease activity.
KEY MESSAGE
1. Certolizumab pegol treatment combined with methotrexate (MTX) in patients with active rheumatoid arthritis and an inadequate response to MTX resulted in rapid onset of efficacy.
2. Rapid onset of efficacy was associated with better clinical outcome at week 24, even in an intermediate tuberculosis (TB)-burden population with adequate TB prophylaxis management.
Notes
We cordially appreciate the support of Marion Boehnlein from UCB. This clinical trial was fully funded by Korea Otsuka Pharmaceutical Co. Ltd., the sponsor of this study.