Small RNA sequencing profiles of mir-181 and mir-221, the most relevant microRNAs in acute myeloid leukemia
Article information
Abstract
Background/Aims
To evaluate and select microRNAs relevant to acute myeloid leukemia (AML) pathogenesis, we analyzed differential microRNA expression by quantitative small RNA next-generation sequencing using duplicate marrow samples from individual AML patients.
Methods
For this study, we obtained paired marrow samples at two different time points (initial diagnosis and first complete remission status) in patients with AML. Bone marrow microRNAs were profiled by next-generation small RNA sequencing. Quantification of microRNA expression was performed by counting aligned reads to microRNA genes.
Results
Among 38 samples (32 paired samples from 16 AML patients and 6 normal marrow controls), 27 were eligible for sequencing. Small RNA sequencing showed that 12 microRNAs were selectively expressed at higher levels in AML patients than in normal controls. Among these 12 microRNAs, mir-181, mir-221, and mir-3154 were more highly expressed at initial AML diagnosis as compared to first complete remission. Significant correlations were found between higher expression levels of mir-221, mir-146, and mir-155 and higher marrow blast counts.
Conclusions
Our results demonstrate that mir-221 and mir-181 are selectively enriched in AML marrow and reflect disease activity. mir-3154 is a novel microRNA that is relevant to AML but needs further validation.
INTRODUCTION
Acute myeloid leukemia (AML) is a group of hematopoietic neoplasms featuring impaired hematopoiesis and bone marrow failure caused by clonal expansion of undifferentiated myeloid precursors. AMLs are frequently found to have recurrent chromosomal aberrations and gene mutations [1]. Even in AML cases with normal cytogenetics, most carry driver mutations relevant to patient clinical outcomes [1]. In the context of posttranscriptional regulation, microRNAs, a class of small noncoding RNAs, regulate the expression of other genes by targeting over 60% of protein-coding transcripts [2].
Compelling evidence has indicated that microRNAs are key regulators of hematopoiesis [3,4]. In AML, several studies have shown that differential microRNA expression is associated with cytogenetic aberrations and impacts clinical outcomes [5-8]. Recently, there has been growing interest in using next-generation sequencing (NGS) to profile microRNAs. Sequencing provides us with the potential to verify novel microRNAs associated with a disease of interest. However, quantitative microRNA sequencing has not been well-evaluated in AML.
In the present study, we analyzed differential microRNA expression by quantitative small RNA NGS using duplicate marrow samples from individual AML patients, and evaluated those microRNAs relevant to AML pathogenesis.
METHODS
Patient samples
All clinical samples included in this study were obtained from AML patients ≥ 20 years of age who underwent bone marrow biopsy before treatment; informed consent was obtained in accordance with the Declaration of Helsinki. Regarding paired samples, we obtained marrow samples at two different time points: initial diagnosis and first complete remission status. Control samples (normal marrow) were collected from the five patients who underwent bone marrow biopsy to rule out primary hematologic disorders and were found normal upon marrow evaluation. The study protocol was approved by the Institutional Review Board at Seoul National University Hospital (1106-092-366).
Total RNA isolation
Briefly, total RNA from marrow mononuclear cells was isolated using Trizol reagent (Invitrogen, Paisley, UK) according to manufacturer instructions. RNA quality was analyzed with a Bioanalyzer and RNA Nano Chips (Agilent Technologies, Santa Clara, CA, USA). Quantification of total RNA yield was determined using QuantiT RiboGreen RNA Reagent and Kit (RiboGreen, Invitrogen).
Small RNA library preparation and sequencing
NGS was performed by LAS Inc. (Gimpo, Korea; http://www.lascience.co.kr/). Small RNAs were sequenced using a TruSeq Small RNA Sequencing Kit (Illumina, San Diego, CA, USA) according to manufacturer instructions. All samples were sequenced on an Illumina NextSeq 500 Sequencer using a 75-cycle High Output Kit.
With the binary base call files from the sequencer, conversation into FASTQ format and de-multiplexing were performed using Illumina bcl2fastq2 software version 2.17 (http://support.illumina.com/downloads/). Adaptor trimming was performed using the FASTQ Toolkit App version 1.0 of Illumna BaseSpace (http://basespace.illumina.com/apps/). Quality of the sequenced reads before and after adapter trimming was evaluated using FastQC software version 0.11.4 (http//www.bioinformatics.babraham.ac.uk/projects/fastqc/). Cleaned sequences were aligned to the most recent mirBASE database release 21 (http://mirBase.org/) [9] using the Small RNA App version 1.0 of Illumina BaseSpace. Quantification of microRNA expression was performed by counting aligned reads to microRNA genes.
Statistical analysis
To identify potential microRNAs that were the most relevant to AML pathogenesis, a three-step approach was employed as follows:
Step 1: comparison of microRNA expression between 14 AML patients and 5 normal marrow controls (Fig. 1A).

Preplanned comparison for differential microRNA (miRNA) expression. (A) Acute myeloid leukemia (AML)-related miRNA, (B) paired sample analysis, and (C) leukemia burden-related miRNA.
Step 2: comparison of microRNA expression using paired samples from the time of diagnosis and complete remission for each patient (Fig. 1B).
Step 3: testing of the association of microRNA expression with different counts of bone marrow blasts (Fig. 1C).
Statistical analyses were completed using Stata version 12.0 software (StataCorp LP, College Station, TX, USA). A Student t test was used to determine statistically significant differences between comparison groups. p values of less than 0.05 were considered statistically significant. Pearson’s correlation coefficient was calculated and analyzed to evaluate the linear relationship between the expression of a specific microRNA and disease burden (the lowest and the highest blast counts in bone marrow).
RESULTS
From March 2013 to August 2015, we enrolled 16 AML patients whose paired samples from the time points of diagnosis and complete remission were available as well as six patients who were confirmed to have normal marrow. Among the 16 paired samples from AML marrow, eight were eligible for analysis after evaluating the quality of extracted RNA. Of the remaining eight paired samples, six single samples obtained at diagnosis were included in further analyses. Among the six samples from normal marrow, five were enrolled as normal controls except one which RNA was not qualified for sequencing; four patients were diagnosed with immune thrombocytopenia and one was diagnosed with normal reactive marrow.
The clinical and molecular characteristics of enrolled patients are shown in Table 1. The median age of the 14 AML patients was 53 years (range, 20 to 78) and seven (50%) were men. Eight patients (57%) had normal karyotypes.
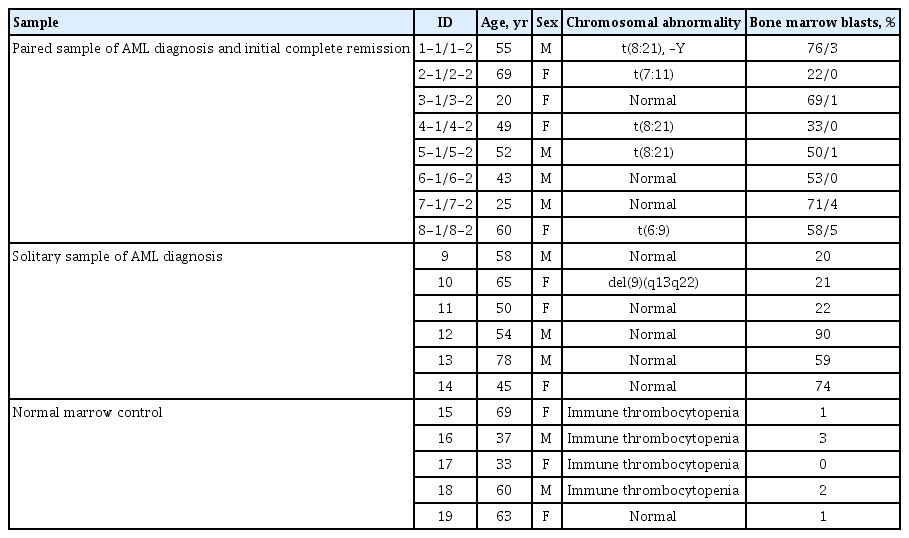
Clinical characteristics of study patients
Descriptive statistics of microRNA differential expression
The number of reads that mapped to alleged microRNAs in the miRBase database ranged from 2,816,113 to 13,405,453. All microRNA mapping reads belonged to 511 unique microRNA families.
Twelve microRNAs were identified as AML relevant microRNA
To establish whether the microRNA profiles of AML cells and normal controls may differ, we first compared the 14 AML samples and five normal controls (Fig. 1A). Upon initial comparison, 19 microRNAs were found to be differentially expressed in AML as compared to normal controls. Twelve of these microRNAs had higher expression levels in AML than in normal controls (Table 2).

AML-related miRNA in 3-comparison approaches
Six microRNAs were related to leukemia activity
Approximately 80% of patients with AML achieve complete remission with conventional induction chemotherapy [10]. In complete remission, the total body leukemic cell population is reduced from approximately 1012 to below approximately 109 cells. Likewise, we hypothesized that expression of AML-relevant microRNAs could substantially decrease from the time of initial diagnosis to remission. Importantly, we assert that using a paired sample from the same patient can minimize interpersonal variation. Therefore, we compared the expression of microRNA between initial diagnosis and complete remission in eight AML paired samples (Fig. 1B). Six microRNAs had higher expression levels at initial AML diagnosis as compared to initial complete remission (Table 2).
Four microRNAs according to AML tumor burden
In the context of the third comparison (Fig. 1C), we assert that certain microRNAs correlated with marrow blasts counts may be relevant to AML. Indeed, we detected positive correlations between higher expression levels of mir-221, mir-146, and mir-155 with higher marrow blast counts, with Pearson’s correlation coefficients of r = 0.680, r = 0.613, and r = 0.592, respectively (Table 2).
DISCUSSION
In the present study, we identified 12 microRNAs that were expressed more highly in AML samples than in normal controls. Among these 12 microRNAs, three were found to reflect AML stage; mir-181, mir-221, and mir-3154 were detected at significantly higher levels upon initial AML diagnosis (before chemotherapy) as compared to complete remission (after chemotherapy). We also explored microRNAs that correlated to marrow blast counts and therefore represented disease burden; high expression levels of three microRNAs (mir-221, mir-146, and mir-155) were significantly correlated with higher marrow blast counts.
Interestingly, all comparisons revealed mir-221 expression levels to be associated with aspects of AML burden. mir-221/222 is known to inhibit normal erythropoiesis and erythroleukemic cell growth [7]. Furthermore, primary microRNA-221/222 is overexpressed in AML, and it is thus a putative oncogene [11,12]. In lymphocytic leukemia, increased expression of mir-221/222 affects the proliferation of leukemic cells [13] and is associated with poorer clinical outcome [14]. We obtained consistent results; mir-221 was expressed significantly higher in AML samples than in normal controls, and mir-221 was downregulated in patients with leukemia remission status after inhibition of proliferation.
A functional relationship exists between mir-221/222 and p27Kip1, a key regulator of the cell cycle; a known target of mir-221/222 includes a regulator of p27Kip1, cyclin- dependent kinase inhibitor 1B (CDKN1B) [15,16]. Regulation of p27Kip1 expression by aberrant mir-221/222 expression promotes cancer cell progression [17]. Thus, the use of synthetic inhibitors of mir-221/222 as a putative oncogene might be a promising approach to cancer treatment [18-20].
The mir-181 family, which was included in comparisons 1 and 2, was the first microRNA family discovered to be specifically expressed in hematopoietic cells [21]. Accumulating data has shown that the mir-181 family plays a critical role in regulating normal cell differentiation and leukemogenesis, particularly in AML [21,22]. A number of studies have shown that mir-181 expression is correlated to cytogenetic and molecular subtypes of AML and has potential as a diagnostic marker for AML [23-25]. Moreover, mir-181 family members have been consistently reported as prognostic markers in AML patients [8,26-28]. Based on these reports, studies on chemotherapy combined with strategies targeting mir-181 have been conducted as a new strategy for AML therapy.
Another microRNA that was included in comparisons 1 and 2, mir-3154, has not been reported in scientific literature to our knowledge. Subsequent studies on the role of mir-3154 for AML pathogenesis are warranted.
Small microRNA sequencing by NGS has many advantages. One clear advantage is the ability to find novel microRNAs. Additionally, because microRNAs have relatively short sequence of 18 to 24 nucleotides, NGS could sequence microRNAs that are vulnerable to rapid degradation [29]. However, there is no evidence that NGS is superior to microarray or real-time polymerase chain reaction [30].
Our findings are limited by selection bias with respect to a 29% sequencing failure rate and small sample sizes. Additionally, the microRNAs reported in the present study need to be validated in independent datasets, and functional characteristics must be elucidated.
Our results demonstrate that mir-221 and mir-181 are selectively enriched in AML marrow and reflect disease activity. A mir-3154 is a novel microRNA that is relevant to AML but requires further validation.
KEY MESSAGE
1. Our study showed that mir-221 and mir-181 are selectively enriched in acute myeloid leukemia (AML) marrow and reflect disease activity.
2. It is meaningful that our results are reproduced in a new methodology (quantitative small RNA next-generation sequencing, duplicate marrow samples from individual AML patients) with small sample size.
3. A mir-3154 is a novel microRNA that is relevant to AML but requires further validation.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2011-0024046 to NRF-2015-0024046), and was supported by the Cooperative Research Program of Basic Medical Science and Clinical Science from Seoul National University College of Medicine (grant no. 800-20140171).