Treatment of diabetic kidney disease: current and future targets
Article information
Abstract
Diabetic kidney disease (DKD) is a leading cause of end-stage renal disease in Korea and worldwide, and is a risk factor for the development of cardiovascular complications. The conventional treatments for DKD are control of blood glucose and blood pressure levels by inhibiting the renin-angiotensin system. However, the prevalence of DKD continues to increase and additional therapies are required to prevent or ameliorate the condition. Many drugs have been, or are being, developed to target the molecular mechanisms in play in DKD. This review focuses on DVD treatment, considering current and emerging therapeutic targets and the clinical trial-based evidence.
INTRODUCTION
Diabetic kidney disease (DKD) is a risk factor for cardiovascular disease and is a leading cause of end-stage renal disease (ESRD) in Korea and worldwide [1]. The Korea National Health and Nutrition Examination Survey V found that the prevalence of albuminuria (defined as a spot urine albumin/creatinine ratio > 30 mg/g) was 26.7% and that of chronic kidney disease (CKD) (defined by a glomerular filtration rate [GFR] < 60 mL/min/1.73 m2) was 8.6% [2]. Given that the number of patients with diabetes is increasing, the incidence of DKD will also rise [1]. A United States report on trends in the incidence of diabetes-related complications between 1990 and 2010 found that ESRD declined less than did acute myocardial infarction and stroke [3]. In addition, the prevalence of albuminuria in diabetic Asian males was higher than that in diabetic European males [4,5]. Therefore, additional efforts are needed to prevent DKD and to delay disease progression. This review considers both current treatments for DKD and some promising new molecular targets.
TREATMENTS FOR DIABETIC KIDNEY DISEASE
Many factors initiate or promote DKD. Initiators include hyperglycemia and altered expression of certain genes. Promoters include hyperglycemia, hypertension, dyslipidemia, smoking, ethnicity, sex, age, and a long diabetes duration. The aim is to target modifiable initiators and promoters [6,7].
CONTROL OF GLUCOSE LEVELS
A great deal of evidence indicates that good control of blood glucose levels prevents DKD development [8]. The Diabetes Control and Complications Trial exploring type 1 diabetes [9], the UK Prospective Diabetes Study (UKPDS) [10], and the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial [11] exploring type 2 diabetes, revealed that intensive therapy reduced the risk of development of diabetic microvascular complications, including nephropathy. The UKPDS trial enrolled patients with newly diagnosed type 2 diabetes and identified a direct relationship between the risk of diabetes-related complications and the level of glycemia, with no “safe” glycemia threshold being defined [12]. However, the ADVANCE trial that enrolled patients with established type 2 diabetes found a non-linear relationship between the hemoglobin A1c (HbA1c) level and the risk of microvascular complications. When the HbA1c level was < 6.5%, there was no evidence of a reduced risk of microvascular complications; however, an HbA1c level > 6.5% was associated with microvascular complications. Indeed, every 1% increase in the HbA1c level was associated with a 40% greater risk of microvascular events [13]. Based on these studies, current guidelines for glycemic control recommend an HbA1c target of 6.5% (Korean Diabetic Association [KDA]) [14] or 7.0% (American Diabetic Association [ADA]) [15] and the Kidney Disease Outcomes Quality Initiative (KDOQI) [16], in the absence of hypoglycemia.
BEYOND GLYCEMIC CONTROL: THE USE OF ORAL HYPERGLYCEMIC AGENTS
Recently, growing evidence suggests that two oral hyperglycemic agents, dipeptidyl peptidase 4 (DPP-4) inhibitors [17,18], and sodium-glucose cotransporter 2 (SGLT2) inhibitors, exert renoprotective effects [19,20].
DPP-4 inhibitors
DPP-4 inhibitors have been widely used since 2006 to treat patients with type 2 diabetes; nine different drugs are licensed in Korea. They inhibit the DPP-4 enzyme, increasing the level of endogenous plasma glucagon-like peptide-1, which promotes insulin secretion but inhibits glucagon secretion; thereby, lowering the blood glucose level. DPP-4 also cleaves substrates such as neuropeptide Y, high-mobility group protein B1, and meprin-β [8,21]. Recent studies suggest that DPP-4 inhibition is associated with pleiotropic effects on cardiorenal protection. Apart from the glucose-lowering effect, the inhibitors exert anti-oxidant, anti-inflammatory, and anti-fibrotic effects [18,21]. DPP-4 is expressed in most organs and cell types, but the activity levels are tissue-dependent. DPP-4 is expressed at relatively high concentrations in the kidney, especially in the brush borders of proximal tubular cells [22,23]. Some studies have found that DPP-4 expression increased in pathological conditions such as diabetic nephropathy [24-26]. Numerous preclinical and clinical studies now support the notion that DPP-4 exerts a renoprotective effect [24,27-38]; the studies are summarized in Table 1.
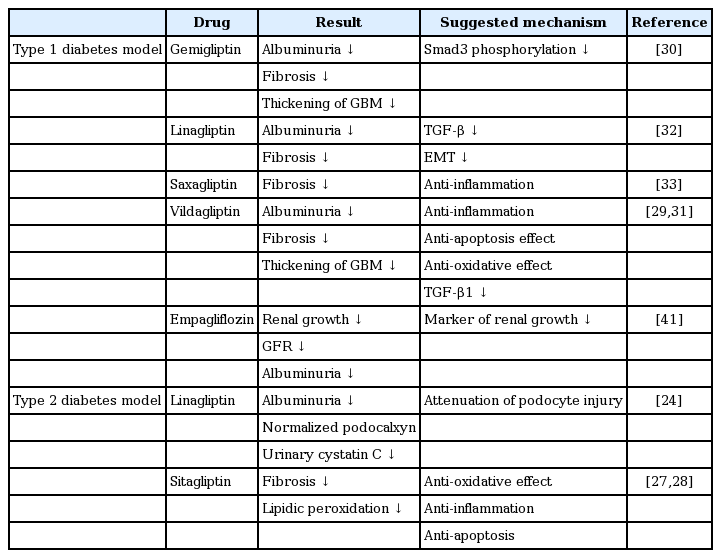
Preclinical studies of renoprotection by DPP-4 and SGLT2 inhibitors
In an animal model of type 2 diabetes, sitagliptin ameliorated diabetic nephropathy at the histological level, exerting anti-oxidant and anti-inflammatory effects [24,27,28]. In a mouse model of type 1 diabetes, DPP-4 inhibitors reduced the extent of albuminuria and attenuated renal fibrosis [29-33]. These studies suggest, therefore, that DPP-4 inhibitors exert renoprotective effects via anti-oxidant and anti-inflammatory mechanisms, and by suppressing transforming growth factor (TGF)-mediated signaling. However, further studies are required.
We also observed, in clinical studies, that DPP-4 inhibition exerted a beneficial effect on the kidney. In a small study, sitagliptin significantly reduced albuminuria without lowering the estimated GFR (eGFR) [34], and alogliptin ameliorated albuminuria in patients with type 2 diabetes by upregulating stromal cell-derived factor-1α and reducing oxidative stress [35]. Two other recent clinical studies also reported reduced albuminuria in patients at the end of treatment [36,37], but did not completely exclude the possibility that this was attributable to the glucose-lowering effect of DPP-4 inhibition. However, preclinical data showing that DPP-4 inhibition exerted a renoprotective effect, independently of the glucose-lowering action, have been corroborated in later clinical studies. Groop et al. [38] reported that linagliptin treatment, coupled with stable inhibition of the renal-angiotensin-aldosterone system, significantly reduced albuminuria in patients with diabetes, and that this was not attributable to a reduced blood glucose level or a lowering of blood pressure (BP) [36]. Additional clinical trials have been completed recently (NCT01792518) or are ongoing (NCT01897532); these trials explore the renal effects of DPP-4 inhibitors using outcome measurements including albuminuria and the eGFR. The studies will provide more information on the effects of DPP-4 inhibition on DKD.
SGLT2 inhibitors
SGLT2 inhibitors are oral hypoglycemic agents that reduce renal glucose re-absorption; thereby, increasing urinary glucose excretion, reducing hyperglycemia. These drugs also lower BP and reduce body weight [39]. A recent study found that empagliflozin retarded the development of kidney disease [40]. The cited work also explored the effects of empagliflozin on microvascular outcomes, especially kidney disease progression, in patients enrolled in a randomized, placebo-controlled cardiovascular outcome trial (EMPA-REG OUTCOME [Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients]). The empagliflozin-treated group exhibited a 39% reduction (compared with placebo) in a composite outcome (de novo or worsening nephropathy, or cardiovascular death). In addition, both the number of patients progressing to macroalbuminuria and those exhibiting a doubling of the serum creatinine level were lower in the empagliflozin-treated group than the placebo group [40].
BP and obesity are also risk factors for DKD [7]; thus, reductions in BP and/or body weight may be beneficial. Inhibition of SGLT2 may trigger such effects. SGLT2 inhibitors prevent the development of glomerular hyperfiltration, an early sign of DKD [41,42]. In addition, any drug-mediated effects on (for example) arterial stiffness and/or vascular resistance would reduce the BP [43]; lowered serum uric acid levels may also retard renal disease progression [20,40]. Currently, the CREDENCE (Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation) trial is evaluating the effects of canagliflozin on renal and vascular outcomes in patients with type 2 diabetes and stage 2 or 3 CKD accompanied by macroalbuminuria (trial code NCT0206579).
CONTROL OF BLOOD PRESSURE
BP control is generally recommended to prevent stroke, cardiovascular disease, and albuminuria. Many studies have shown that BP control is renoprotective [8]. The UKPDS suggested that a 10-mmHg decrease in systolic BP was associated with reduced levels of diabetic microvascular complications, including nephropathy [44]. The ADVANCE study found that a 5.6-mmHg fall in systolic BP reduced the risk of major macro- or microvascular events; in particular, the development of microalbuminuria was significantly reduced [45]. Therefore, to prevent the development and progression of DKD, the ADA recommends that treatment should aim to reduce the BP below 140/90 mmHg [46]. The KDOQI guidelines recommend that adults with diabetes, but without albuminuria, should be treated to maintain a systolic BP consistently ≤ 140 mmHg and a diastolic BP consistently ≤ 90 mmHg, whereas adults with albuminuria should maintain a systolic BP that is consistently ≤ 130 mmHg and a diastolic BP that is consistently ≤ 80 mmHg [47]. Finally, the KDA recommends that the BP should be held at < 140/85 mmHg [14].
Angiotensin II receptor blockers (ARBs) or angiotensin-converting enzyme (ACE) inhibitors are recommended to control BP [14,46,47]. Many trials have shown that ARBs or ACE inhibitors delay ESRD progression and development [48]. However, combination treatment with an ARB and an ACE inhibitor is not recommended because of the lack of evidence for any beneficial effect on cardiovascular disease or DKD over and above onedrug treatment, and the higher prevalence of adverse events such as hyperkalemia [49].
NOVEL DRUGS FOR THE TREATMENT OF DIABETIC KIDNEY DISEASE
Even when the glucose level and BP are controlled, some diabetes patients still progress to ESRD. Therefore, additional preventative strategies are needed. Several trials using novel drugs targeting the molecular mechanisms of ESRD development have recently been completed or are ongoing (Fig. 1).
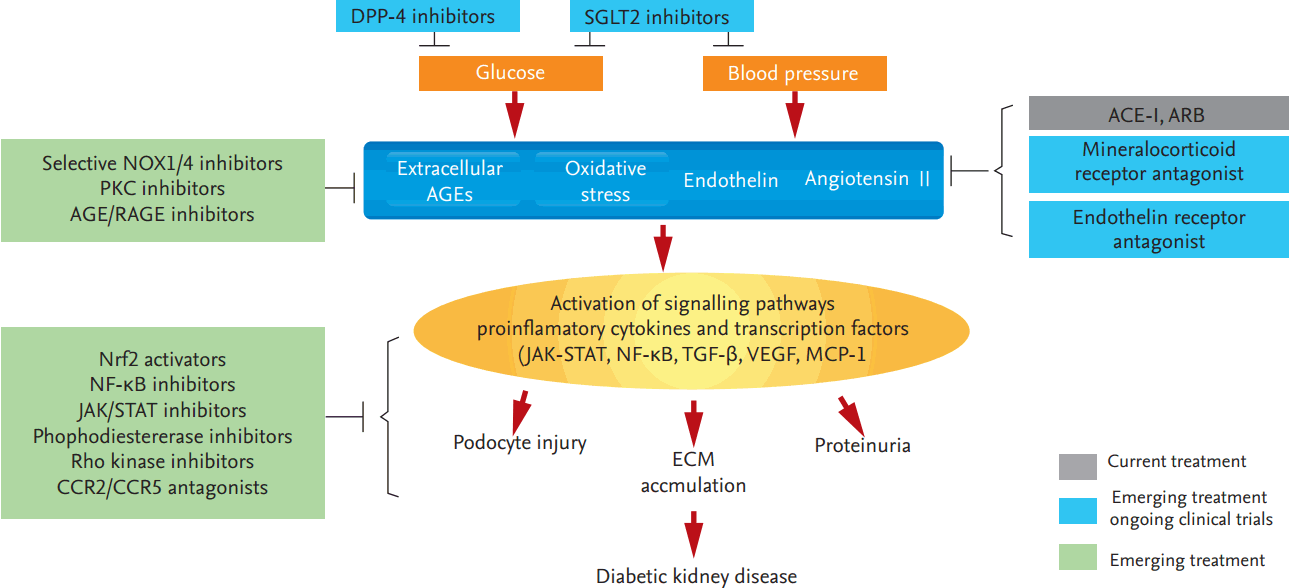
Current and emerging treatments for diabetic kidney disease. DPP-4, dipeptidyl peptidase-4; SGLT2, sodium-glucose cotransporter 2; NOX, NADPH oxidase; PKC, protein kinase C; AGE, advanced glycation endproduct; RAGE, receptor for advanced glycation endproduct; ACE-I, angiotensin converting enzyme-inhibitor; ARB, angiotensin II receptor blocker; Nrf2, nuclear factor-like 2; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; JAK/STAT, Janus kinase-signal transducer and activator transcription factor; CCR, C-C chemokine receptor; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor; MCP-1, monocyte chemotactic protein 1; ECM, extracellular matrix.
Mineralocorticoid receptor antagonists
Aldosterone activates the mineralocorticoid receptor to regulate the sodium balance, but also promotes inflammation and fibrosis [50]. The mineralocorticoid receptor antagonists (MRAs) spironolactone and eplerenone increase the risk of hyperkalemia in patients with diabetes and CKD [51]. Patients with CKD and heart failure treated with the non-steroidal MRA finerenone (BAY 94-8862) exhibited reduced albuminuria, which was associated with a lower risk of hyperkalemia than was spironolactone [52]. In addition, a phase II clinical trial explored the utility of finerenone in patients with diabetic nephropathy who were also receiving an ARB or an ACE inhibitor. In such patients, finerenone reduced albuminuria in a dose-dependent manner; however, 1.8% of patients receiving the drug, but none in the placebo group, developed hyperkalemia; no other adverse effect differed in prevalence between the groups [53]. The drug is now in an ongoing phase III trial (code NCT02540993).
Endothelin receptor antagonists
Endothelin 1 (ET-1) is a powerful vasoconstrictor and mitogenic factor exhibiting vasoactive, inflammatory, and profibrogenic properties, and is implicated in cardiovascular and CKD. ET-1 contributes to renal fibrosis via various mechanisms, promoting the accumulation of extracellular matrix components and endothelial cell proliferation, stimulating the epithelial-mesenchymal transition, and upregulating the production of cytokines and growth factors [54]. In a phase III clinical trial, an ET receptor antagonist, avosentan, reduced albuminuria. However, the trial was terminated prematurely because the drug significantly increased fluid overload and the incidence of congestive heart failure. Such adverse effects may be attributable to a natriuretic effect of ETB receptor inhibition [55]. A clinical trial of the highly selective ETA receptor antagonist, atrasentan, is ongoing (code NCT01858532).
Apart from these drugs, further materials targeting additional mechanisms linked to DKD progression, such as oxidative stress and uric acid metabolism, are under development [56].
EMERGING THERAPEUTIC TARGETS
Autophagy
Autophagy is the process by which mammalian cells degrade their own macromolecules and organelles to maintain intracellular homeostasis, and it protects organisms against diverse pathologies, including infections, cancers, neurodegeneration, aging, and heart disease [57]. Recently, autophagy has been implicated in the pathology of a number of diseases, including cancer and diabetes [58,59]. However, accumulating evidence suggests that autophagy plays a cytoprotective role in the kidney [60,61]. Renal autophagy is activated under certain stress conditions, including oxidative stress and hypoxia, and in kidneys of diabetic animal models, but is suppressed in obese patients with type 2 diabetes [62,63]. Autophagy has been observed both in podocytes and proximal tubular cells, but plays contrasting roles in these cell types. Podocytes require a basal level of autophagy to maintain cellular homeostasis, whereas proximal tubular cells use autophagy as a coping mechanism when nephrotoxic stress is in play. Therefore, if a treatment for diabetic nephropathy were to seek to regulate autophagy, the specificity of such treatment might be problematic [60].
Src family kinases
The Src family kinases (SFKs) are non-receptor tyrosine kinases activated by autophosphorylation of their Tyr416 residues, and are induced by certain cytokines and growth factors including TGF-β1 and epidermal growth factor [64]. The SFKs include Src, Fyn, Yes, Yrk, Blk, Fgr, Hck, Lck, Lyn, and the Frk subfamily proteins [65]; all contain a unique amino-terminal region, two Src homology domains (SH2 and SH3), highly conserved kinase domains, and carboxy-terminal tails containing the (negatively) regulatory tyrosine residue [66]. Src, Fyn, and Yes are expressed in most tissues, but their activities are tissue-dependent; the other SFKs are typically expressed in hematopoietic cells [65]. SFKs dysregulate many cellular processes, including invasion, migration, proliferation, angiogenesis, and apoptosis, and have been targeted principally in efforts to treat various types of cancer [66].
Recent studies have suggested that the Src is a potential target when it is sought to treat kidney fibrosis. High glucose levels activate the Src of mesangial cells and the glomeruli of rats with streptozotocin (STZ)-induced diabetes [67,68]. Taniguchi et al. [68] reported that pharmacological inhibition of Sfc by PP2 and SU6656 blocked high-glucose-stimulated Sfc phosphorylation (at Tyr-416), the EGF receptor (EGFR), mitogen-activated protein kinase, and the tumor necrosis factor α-converting enzyme. Moreover, diabetic STZ-treated mice treated with PP2 exhibited reduced albuminuria, less glomerular collagen accumulation, and reduced podocyte loss, attributable to inhibition of EGFR phosphorylation [68]. Yan et al. [69] reported that PP1-mediated inhibition of the Src kinase reduced fibrosis in either the presence or absence of TGF-β stimulation in vitro, and attenuated renal interstitial fibrosis after the development of unilateral ureteral obstruction in vivo. These renoprotective effects were mediated by suppression of TGF-β signaling, reduced phosphorylation of both EGFR and the signal transducer and activator of transcription-3 (STAT-3), and cell-cycle arrest [69]. Src inhibitors are currently being developed for cancer patients; various treatments are being evaluated in phase 0-to-phase III clinical trials [66]. As SRC inhibition reduces renal fibrosis, Zhou and Liu [70] have proposed that such an effect could translate into effective remedies for patients with kidney disease. Apart from the Src effects, we recently showed that Fyn, another SFK, was activated in animals with unilateral ureteral obstruction-induced renal fibrosis, and that Fyn inhibition attenuated fibrosis by downregulating STAT-3 [71]. Together, the results suggest that SFK may be a valuable therapeutic target in patients with renal fibrosis.
CONCLUSIONS
The incidence of DKD is expected to increase as the incidence of diabetes rises. Although control of glucose and BP levels sometimes prevent DKD development, many patients with diabetes progress to ESRD. DKD development and progression are associated with numerous modifiable and non-modifiable risk factors. Therefore, to treat DKD more effectively, it is essential to identify novel drug targets based on better knowledge of DKD pathogenesis, and to judge the success of such therapies using reliable markers of DKD and ESRD progression.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2015R1C1A2A01052054).