A novel c.563 T>G, p.L189R lamin A/C mutation in identical twins with dilated cardiomyopathy
Article information
To the Editor,
Dilated cardiomyopathy (DCM) presents with left or both left ventricular (LV) and right ventricular (RV) dilatation, and reduced systolic function. The etiologies of DCM are varied, with about half of cases considered idiopathic and 20% to 50% considered familial. More than 60 genes, including lamin A/C gene (LMNA), were found to be related to familial DCM. We report a case of identical twins with DCM who had the same gene mutation but different phenotypes.
A 30-year-old man was referred to us for the management of heart failure (index patient). He had a history of atrial fibrillation diagnosed approximately 10 years ago. However, he had been asymptomatic with adequate exercise capacity and had not received treatment. One month before the admission, he developed shortness of breath, productive cough, and blood-tinged sputum. He was diagnosed with congestive heart failure and started treatment with β-blocker, angiotensin-converting enzyme inhibitor, digoxin, aspirin, and direct thrombin inhibitor. Then, he was transferred to our hospital for further evaluation and management. He was of normal weight, and a nonsmoker and nonalcoholic. His general feature and physical examination results were normal, except for an irregular heart rhythm. Electrocardiography (ECG) revealed atrial fibrillation with premature ventricular contractions (Fig. 1A). Transthoracic echocardiography revealed LV dilation with reduced LV systolic function (LV end-diastolic dimension, 56 mm; ejection fraction, 28%), markedly enlarged left atrium (left atrial volume index, 70 mL/m2) (Fig. 1B), and mild resting pulmonary hypertension (estimated pulmonary artery systolic pressure, 46 mmHg) with RV dysfunction. To further characterize the myocardium, cardiac magnetic resonance imaging (MRI) and endomyocardial biopsy were performed. Cine MRI confirmed markedly increased LV volume and the presence of LV systolic dysfunction (LV end-diastolic volume, 213 mL; ejection fraction, 17%). Late gadolinium-enhanced MRI demonstrated linear midwall fibrosis, which is commonly found in idiopathic DCM (Fig. 1C). Endomyocardial biopsy revealed hypertrophy of cardiac myofibers, mild subendocardial fibrosis, and minimal lymphocytic infiltration in the interstitium. No evidence of infiltrative cardiomyopathy, including amyloidosis and sarcoidosis, or features of vasculitis were observed.
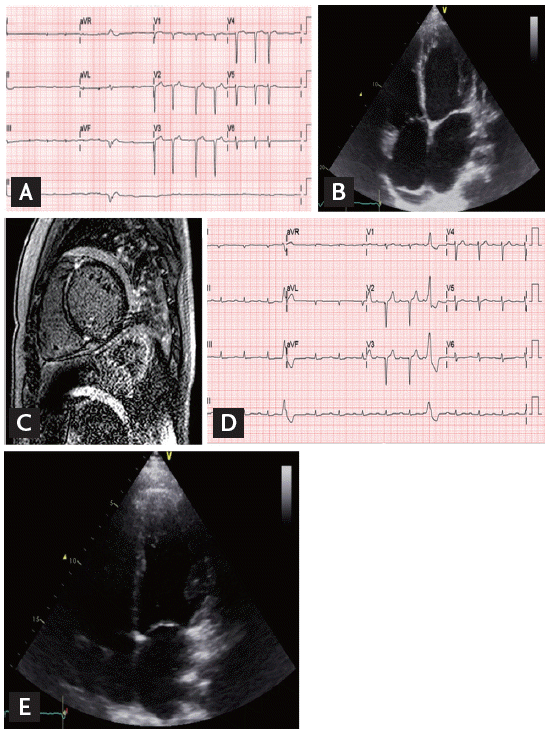
The images of the index patient (A-C) and his identical twin (D, E). (A) The electrocardiography (ECG) of index patient shows atrial fibrillation and diffuse T-wave flattening and low QRS amplitude. (B) The transthoracic echocardiography (TTE) shows left ventricular (LV) dilatation and LV enlargement. (C) The T2-weighted image shows no evidence of definite high signal intensity. (D) The ECG of identical twin maintains sinus rhythm but the first degree atrioventricular block, low QRS amplitude and ventricular premature beats are also present. (E) Despite no symptoms, his TTE finding is similar to the index patient.
Early-onset DCM with arrhythmia raised the clinical suspicion of familial DCM. The pedigree is illustrated in Fig. 2. The patient’s father was diagnosed with heart failure with conduction abnormality in his thirties and was scheduled to undergo pacemaker implantation. However, he died from decompensated heart failure shortly after diagnosis. The patient’s uncle also died in his thirties from an undefined cardiac disease. Because he had an identical twin brother who were raised in the same family, we also performed clinical evaluation for his identical twin. He was asymptomatic and had no heart failure symptoms. He was also of normal weight, and a nonalcoholic and nonsmoker. His general feature and physical examination results were unremarkable, except for an irregular pulse. The results of all laboratory examinations were within the normal ranges. ECG revealed sinus rhythm, first-degree atrioventricular (AV) block, and ventricular premature beats (Fig. 1D). Although he was asymptomatic, transthoracic echocardiography revealed LV dilatation and systolic dysfunction (LV end-diastolic dimension, 54 mm; ejection fraction, 31%). The LA was also enlarged (left atrial volume index, 44.7 mL/m2) (Fig. 1E).
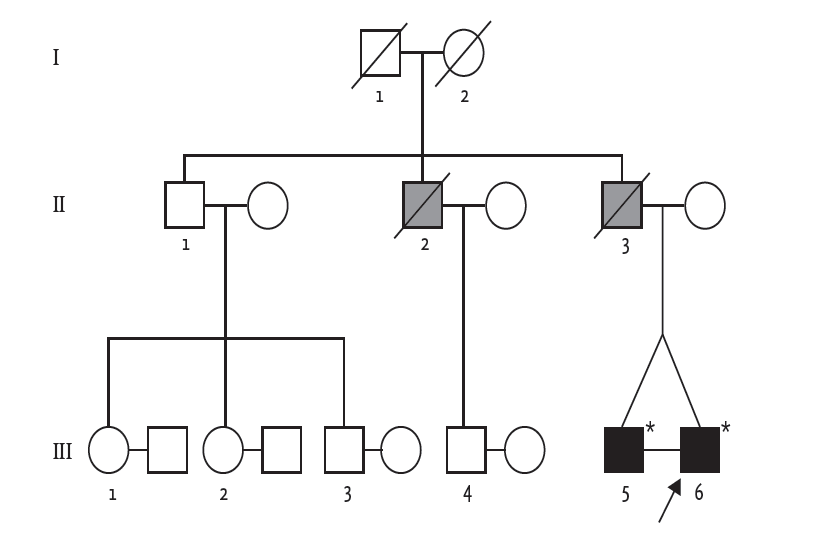
The pedigree of the family. The arrow indicates the index patient. The gray-filled square represents a family member who died of sudden cardiac death. The asterisk represents a genotyped family member.
We performed a genetic screening for DCM by using a multigene panel consisted of in 38 known DCM-related genes. Among the variants detected, all previously reported mutations and probable pathogenic variants including novel nonsynonymous ones were confirmed by Sanger sequencing, as well as all low-coverage regions with coverage depth under 10X. As a results, a novel missense mutation in the LMNA gene, c.563T>G (p.Leu188Arg), was confirmed in both twins (reference cDNA sequence, NM_170707.3) (Fig. 3). Considering that LMNA-related DCM has been known to be a high-risk factor of fatal arrhythmic complications and is prone to lead to sudden cardiac death (SCD), an implantable cardioverter-defibrillator (ICD) was implanted in the twins. The index patient was treated with β-blocker, angiotensin receptor blocker, diuretics, and direct thrombin inhibitor, and was discharged without heart failure symptoms. However, a year later, he presented with dyspnea and lower extremity edema, and was hospitalized for heart failure. His cardiac rhythm indicated atrial fibrillation, but a few events of nonsustained ventricular tachycardia were recorded in ICD. He was admitted for heart failure twice in 1 year despite intensive medical treatment. He also developed RV failure. Finally, he underwent heart transplantation.
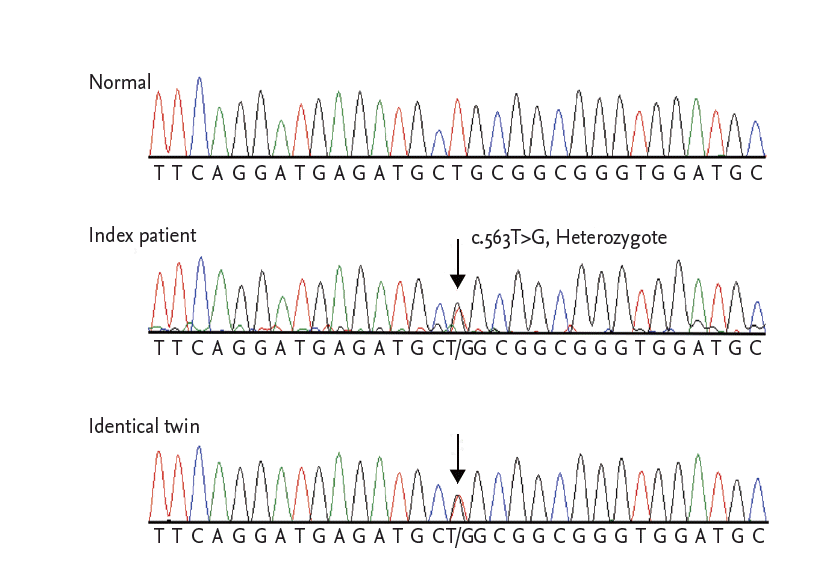
Sequence analysis of LMNA gene. The index patient is heterozygous for the missense variant, c.563T>G. The variant was also detected in his brother.
Although the identical twin was asymptomatic at the time of diagnosis, he started treatment with a β-blocker and an angiotensin-converting enzyme inhibitor. Because he maintained sinus rhythm, anticoagulation treatment was not administered. However, 15 months after the diagnosis, he presented with flank pain and was diagnosed with embolic renal infarction. In his visit to the emergency department, ECG demonstrated atrial fibrillation, which spontaneously converted to sinus rhythm after a day. Transesophageal echocardiography revealed left atrial appendage thrombus. Anticoagulation was started. Despite the event, he remained in good condition without heart failure symptoms.
During the past decade, the understanding of the genetics of familial DCM has significantly progressed. A consensus has been reached that familial DCM can be expected to occur in at least 20% to 35% of individuals with idiopathic DCM with clinical screening of first-degree family members [1]. Mutations in the LMNA gene are the most frequent cause of familial DCM found in 6% of patients presenting with familial DCM [1]. The mode of inheritance of the mutation in DCM is mainly autosomal dominant. The LMNA is expressed in chromosome 1q21.2-q21.3; encodes lamin A and C proteins, which are located beneath the inner nuclear membrane; and composed of a meshwork that provide structural integrity to the nucleus [2]. The proteins are expressed in various tissues, including muscles, nervous tissues, and adipose tissues. Therefore LMNA mutation involves not only the myocardium and conduction systems but also the skeletal muscles, nervous systems, and adipose tissues [2].
LMNA-related DCM usually presents in early to mid-adulthood with symptomatic conduction system disease or arrhythmias, followed by LV dilatation and dysfunction stages [3]. Advanced forms of dysrhythmia such as supraventricular or ventricular tachyarrhythmia develop frequently, and SCD can occur, in some instances, as the presenting manifestation even with preserved LV systolic dysfunction [4]. Therefore, ICD implantation is indicated in patients with LMNA mutation and conduction disorders [5]. Heart failure usually becomes apparent a few years after the onset of dysrhythmia, and the prevalence of heart failure is lower than that of dysrhythmia [3]. The severity of heart failure in each carrier varies from a subclinical state to end-stage heart failure. Standard heart failure treatment, including angiotensin-converting enzyme inhibitor and β-blocker, is applied in symptomatic patients. Meanwhile, cardiac transplantation is the treatment option for end-stage DCM [3].
In this letter, we present identical twins with familial DCM who were found to have a novel functional c.563T>G, p.L188R LMNA mutation. Residue was located in the coil 1B domain, but the impact of the change of the residue has not been fully evaluated. The phenotype of the novel mutation includes dysrhythmia (first-degree AV block, atrial fibrillation, and ventricular arrhythmia) and DCM (LV dilatation, dysfunction, myocardial fibrosis, and combined RV dysfunction). These manifestations are similar to those observed in patients with other LMNA mutations. Interestingly, the identical twins demonstrated the same mutation in LMNA but different clinical manifestations. The index patient was diagnosed with atrial fibrillation in his early twenties and then experienced heart failure 10 years later. His identical twin maintained sinus rhythm and experienced first paroxysmal atrial fibrillation at the age of 32 years but with embolic event as the presenting manifestation. While the index patient underwent heart transplantation because of decompensated heart failure, his identical twin did not show any heart failure symptoms until the age of 32 years even though he also had features of DCM.
In summary, we describe identical twin brothers who carried the novel missense LMNA mutation and found some pathognomonic characteristics of LMNA mutation related to DCM. Interestingly, they had the same mutation but had significantly different clinical courses. ICD was implanted in both brothers to prevent SCD, and heart transplantation was performed in the index patient. Although the identical twin had been free from heart failure symptoms, we should continue to manage him carefully for a long period of time. The importance of genetic counseling regarding the risk of SCD and heart failure harboring LMNA mutations should be emphasized.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
The genetic analysis was supported by the National Supporting Program for Genetic Diagnosis of Rare Diseases of the Korea Centers for Disease Control & Prevention.