Angiotensin III increases monocyte chemoattractant protein-1 expression in cultured human proximal tubular epithelial cells
Article information
Abstract
Background/Aims:
We investigated whether angiotensin III (Ang III) is involved in monocyte recruitment through regulation of the chemokine monocyte chemoattractant protein-1 (MCP-1) in cultured human proximal tubular epithelial cells (HK-2 cells).
Methods:
We measured MCP-1 levels in HK-2 cells that had been treated with various concentrations of Ang III and Ang II type-1 (AT1) receptor antagonists at various time points. The phosphorylation states of p38, c-Jun N-terminal kinases (JNK), and extracellular-signal-regulated kinases were measured in Ang III-treated cells to explore the mitogen-activated protein kinase (MAPK) pathway. MCP-1 levels in HK-2 cell-conditioned media were measured after pre-treatment with the transcription factor inhibitors curcumin or pyrrolidine dithiocarbamate.
Results:
Ang III increased MCP-1 protein production in dose- and time-dependent manners in HK-2 cells, which was inhibited by the AT1 receptor blocker losartan. p38 MAPK activity increased significantly in HK-2 cells exposed to Ang III for 30 minutes, and was sustained at higher levels after 60 minutes (p < 0.05). Total phosphorylated JNK protein levels tended to increase 20 minutes after stimulation with Ang III. Pre-treatment with a p38 inhibitor, a JNK inhibitor, or curcumin significantly inhibited Ang III-induced MCP-1 production.
Conclusions:
Ang III increases MCP-1 synthesis via stimulation of intracellular p38 and JNK MAPK signaling activity and subsequent activated protein-1 transcriptional activity in HK-2 cells.
INTRODUCTION
The renin-angiotensin system (RAS) regulates homeostatic mechanisms such as water and electrolyte balance, blood pressure [1-3], and the aging process [4]. Chronic RAS activation is commonly related to cardiovascular dysfunction and damage to target organs, including the blood vessels, heart, and kidneys [5].
RAS activation causes an inflammatory response by inducing the activities of several cytokines, chemokines, and transcription factors [6,7]. Angiotensin (Ang) II commonly increases the expression of adhesion molecules and cytokines, such as selectins, intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and chemokine (C-C motif) ligand 2/monocyte chemoattractant protein-1 (MCP-1) in target cells, resulting in the recruitment of inflammatory cells from the blood to tissues [8,9].
Metabolic products such as Ang III, Ang IV, and Ang 1 to 7, have been discovered by investigating enzymatic degradation of Ang II in kidney tissues [10]. These products bind to two major receptor subtypes that mediate RAS actions, such as the Ang II type-1 (AT1) and AT2 receptors. However, they also serve as ligands to other receptors, such as the Mas receptor or insulin-regulated aminopeptidase, which have different functions. These receptors have been detected in various regions of the kidney, and their specific roles and functions have been investigated [11-13].
Ang III has similar activity to that of Ang II, and its receptor is located in renal proximal tubular epithelial cells [14,15]. Additionally, aminopeptidase A, present on the surface of podocytes, mesangial cells, and proximal tubular epithelial cells, degrades Ang II into Ang III [16,17]. This degradation product increases the secretion of angiotensinogen and transforming growth factor-β, resulting in vascular contraction and renin secretion, linked to inflammation and fibrosis in various kidney diseases [12,18,19]. However, the details of Ang III function have not been fully elucidated.
MCP-1 is a C-C chemokine with strong chemotactic activity for monocytes, T lymphocytes, and basophils and is expressed by most cells in the kidney, such as mesangial, glomerular epithelial, and proximal tubular epithelial cells [20,21]. In addition, MCP-1 is synthesized and released by vascular, cardiac, and renal cells in response to hemodynamic (shear stress, blood flow, or oxidative stress) and humoral stimuli (such as Ang II and endothelin-1) [22].
Mitogen-activated protein kinases (MAPK) are serine/threonine-specific protein kinases that respond to extracellular stimuli (mitogens, osmotic stress, heat shock, and pro-inflammatory cytokines), and regulate various cellular activities, such as gene expression, mitosis, differentiation, proliferation, and cell survival/apoptosis [23]. p38, extracellular signal-regulated kinases (ERK), and c-Jun N-terminal kinase (JNK) are well-known MAPKs that regulate MCP-1 expression in various kidney diseases, such as proliferative glomerulonephritis and diabetic nephropathy [24-28].
In the present study, we investigated whether Ang III affects MCP-1 expression and activation of transcriptional factors such as nuclear factor kappa B (NF-κB) and activating protein-1 (AP-1) in cultured human proximal tubular HK-2 cells. We also investigated MAPK signaling as an intracellular pathway related to MCP-1 expression by Ang III.
METHODS
Materials
Ang II and III were purchased from Sigma (St. Louis, MO, USA). The primary antibodies used for Western blot were anti-p38, anti-JNK, and anti-ERK rabbit polyclonals (Cell Signaling Technology Inc., Beverly, MA, USA). Inhibitors of p38 MAPK (SB202190), ERK (PD98059), and JNK (SP600125) were purchased from Sigma.
To evaluate the relation of transcription factors to increases in MCP-1, we used the AP-1 inhibitor curcumin (Sigma) and the NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC, Sigma).
Cell cultures
HK-2 cells and immortalized human proximal tubular epithelial cells were purchased from the American Type Culture Collection (Rockville, MD, USA). Cells were seeded at a density of 1 × 104 cells/well into 24-well culture plates containing Dulbecco’s Modified Eagle’s medium-F12 (Gibco BRL, Rockville, MD, USA), supplemented with 2 mM L-glutamine, penicillin (100 U/mL)/streptomycin (100 μg/mL), and 10% fetal bovine serum in humidified air at 37°C with 5% CO2. Medium was changed 48 hours after plating. Cells were cultured to ± 80% confluency, washed twice with HBSS, and starved in serum-free medium for 24 hours [29].
MCP-1 enzyme-linked immunosorbent assay
Mouse anti-human MCP-1 antibody (2 μg/mL; R&D Systems, Minneapolis, MN, USA) was used as the capture antibody, and 1 μg/mL biotinylated goat anti-human MCP-1 antibody (R&D Systems) was used for detection. The TMB micro-well peroxidase substrate system (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD, USA) was used as the enzyme substrate, and the reaction was measured at OD450.
Cell viability
HK-2 cell viability was assessed by measuring lactate dehydrogenase (LDH) release into the culture medium using a LDH cytotoxicity detection kit according to the manufacturer’s protocol (Takara Biomedical, Kyoto, Japan).
Western blot analysis
Confluent cells (1 × 105) were starved for 24 hours and pre-incubated with Ang III (10−7 M) for various times (5, 10, 20, 30, and 60 minutes). Treated cells were lysed for 10 minutes on ice in lysis buffer (50 mM Tris, pH 7.5, 40 mM NaCl, 1% Triton X-100, 2 mM ethylenediaminetetraacetic acid, 1 μg/mL leupeptin, 2 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride). Lysates were cleared by centrifugation at 14,000 ×g (4°C) for 10 minutes. Protein levels were quantified using the Bradford assay. Equal amounts of lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred to Bio-Blot nitrocellulose membranes (Bio-Rad Laboratories Inc., Hercules, CA, USA). Membranes were blocked with TBS (pH 7.6)/5% nonfat dry milk/0.05% Tween 20 and blotted with the indicated antibodies (against phosphorylated and non-phosphorylated p38, JNK, and ERK) at 4°C overnight, then 1:1,000 diluted horseradish peroxidase-conjugated secondary antibody (Amersham Bioscience, Little Chalfont, UK) at room temperature for 1 hour and visualized with an enhanced chemiluminescence kit (Amersham Bioscience).
Statistics
Data are expressed as the mean ± standard error. Multiple groups were compared using the Kruskall-Wallis test. Comparisons between multiple timepoints were performed by repeated-measures analysis of variance. A p < 0.05 was significant.
RESULTS
Ang III increases MCP-1 production in HK-2 cells
Ang III significantly increased MCP-1 protein levels in HK-2 cells in a concentration-dependent manner, which was significantly inhibited by the AT1 receptor antagonist losartan (p < 0.05) (Fig. 1). MCP-1 protein levels were also measured in the supernatants of HK-2 cells stimulated with Ang III (10−7 M) for various time points (8, 12, 24, and 48 hours). Production of MCP-1 by HK-2 cells was significantly stimulated by Ang III after 48 and 72 hours (p < 0.05) (Fig. 2).

Angiotensin III (Ang III)-induced monocyte chemoattractant protein-1 (MCP-1) production in HK-2 cells via the Ang II type-1 (AT1) receptor. HK-2 cells were treated with Ang II (10−6 M) and Ang III (10−9 to 10−6 M) for 48 hours in the presence or absence of the AT1 receptor antagonist losartan (10−7 M). MCP-1 protein in conditioned medium was quantified by enzyme-linked immunosorbent assay. Results are expressed as the percentage increase over untreated cells. Results are shown as mean ± standard error of mean from six independent experiments. ap < 0.05 vs. untreated cells, bp < 0.05 vs. Ang III (10−7 M)-treated cells.
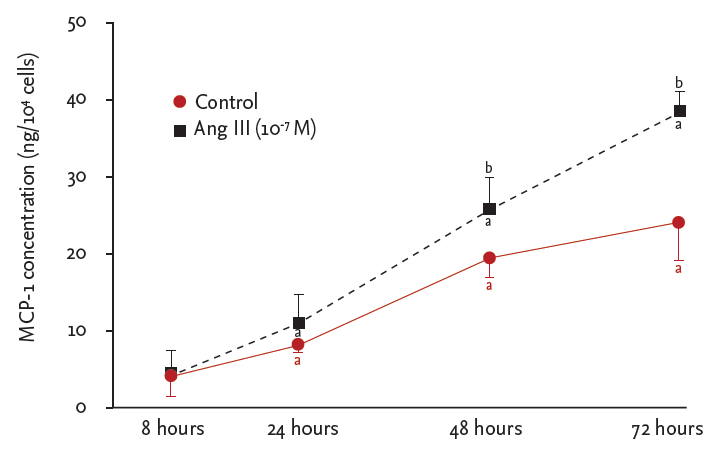
Angiotensin III (Ang III)-stimulated monocyte chemoattractant protein-1 (MCP-1) production in HK-2 cells. Cells were incubated for the indicated times in the presence or absence of Ang III. MCP-1 protein in culture medium was quantified by enzyme-linked immunosorbent assay. Results are shown as mean ± SEM from six independent experiments. Lactate dehydrogenase (LDH) release from Ang II-, Ang III-, or losartan-treated cells. LDH release is expressed as percentage of maximal LDH release induced by 1% Triton X-100 for 48 hours. ap < 0.05 vs. 8 hours MCP-1 level, bp < 0.05 vs, control cells.
LDH release
LDH release did not increase above control values in response to either Ang II (10−6 M), Ang III (10−7 M), or losartan (10−7 M), indicating that these agents are not cytotoxic (Fig. 3).
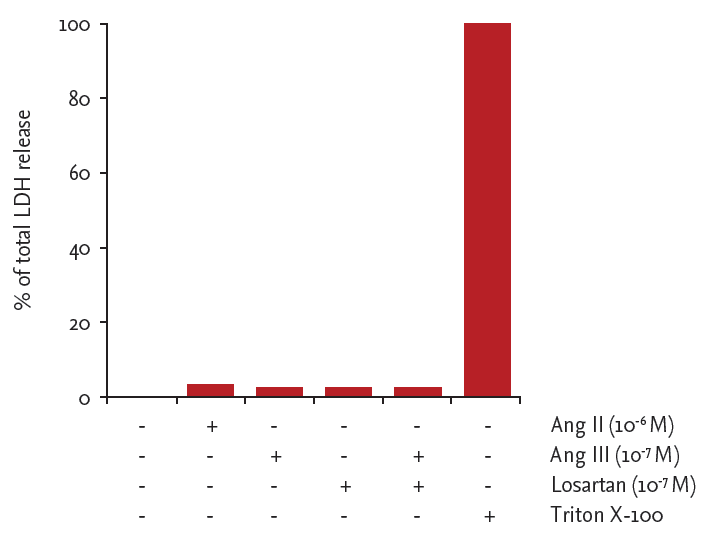
Lactate dehydrogenase (LDH) release from angiotensin (Ang) II-, Ang III-, or losartan-treated cells. LDH release is expressed as percentage of maximal LDH release induced by 1% Triton X-100 for 48 hours.
Ang III stimulates p38 phosphorylation and JNK MAPK activity
To explore whether Ang III induces the MAPK signaling pathway in HK2 cells, the phosphorylation status of p38, JNK, and ERK was measured in Ang III (10−7 M)-treated cells by Western blot using specific antiphospho-MAPK antibodies.
p38 MAPK activity increased significantly in HK-2 cells exposed to Ang III for 30–60 minutes, with peak phosphorylation at 30 minutes (p < 0.05) (Fig. 4A). Total phosphorylated JNK appeared to increase suddenly 20 minutes after Ang III stimulation, but this change was not significant (Fig. 4B). ERK protein levels tended to increase in a time-dependent manner (p > 0.05) (Fig. 4C).
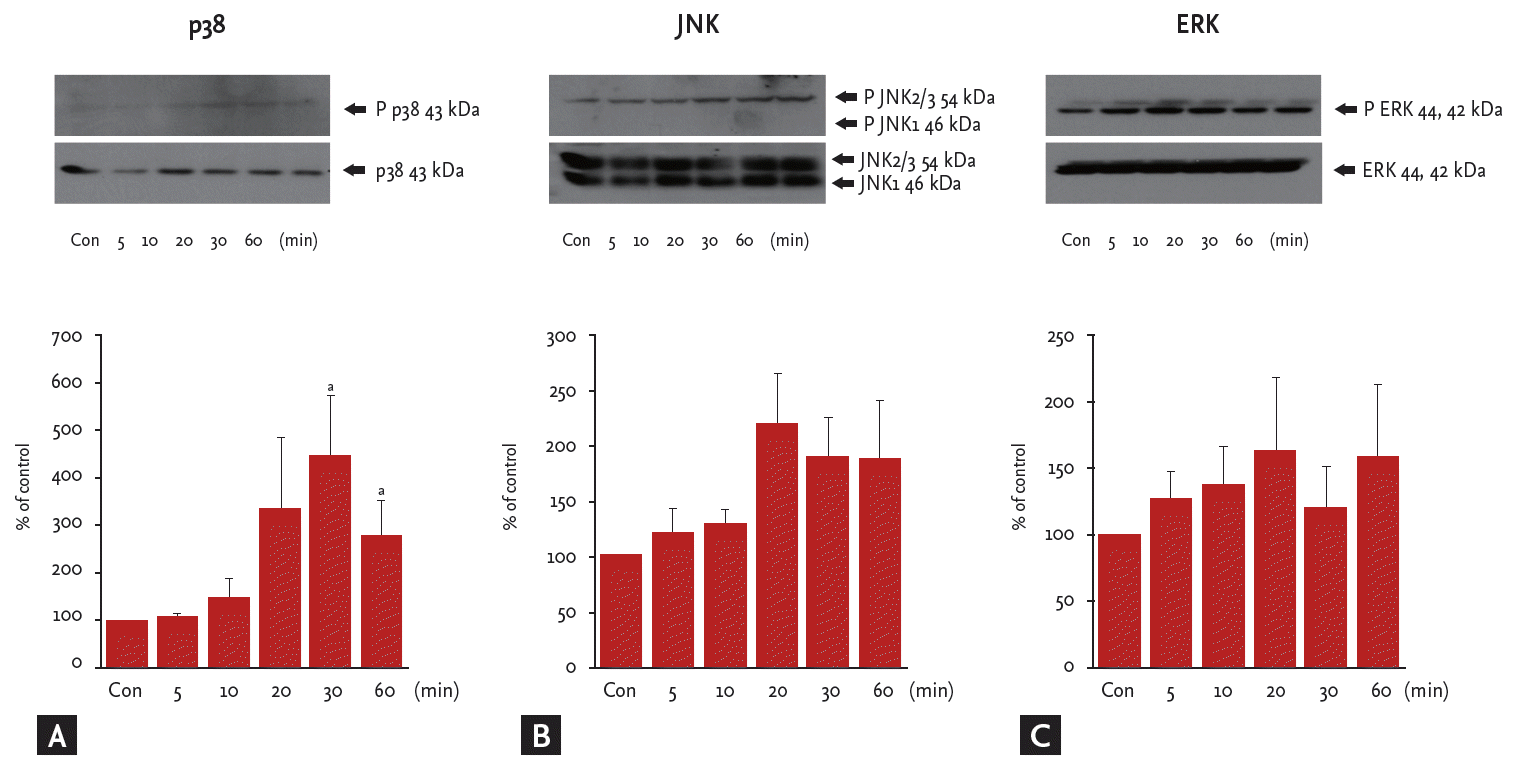
Angiotensin III (Ang III, 10−7M) significantly stimulates p38 phosphorylation. Cells were incubated with Ang III (10−7 M) for various times, and (A) phosphorylated p38, (B) c-Jun N-terminal kinases (JNK), and (C) extracellular signal-regulated kinases (ERK) were detected by Western blot. Results are representative of three independent experiments with similar results. Con, control. ap < 0.05 vs. untreated cells.
To evaluate whether inhibiting various MAPK pathways affects Ang III-induced MCP-1 expression, we measured MCP-1 levels in conditioned media of HK-2 cells pre-incubated with various MAPK inhibitors for 30 minutes and then added Ang III (10−7 M) for 48 hours. Pre-treatment with p38 and JNK inhibitors significantly inhibited Ang III-induced MCP-1 production (p < 0.05) (Fig. 5).

The effect of mitogen-activated protein kinase (MAPK) inhibitors on angiotensin III (Ang III)-induced monocyte chemoattractant protein-1 (MCP-1) production. HK-2 cells were pre-incubated with MAPK inhibitors for 30 minutes and then incubated with Ang III (10−7 M) for 48 hours. MCP-1 protein levels were measured by enzyme-linked immunosorbent assay. Results are expressed as percent increase compared to untreated cells. Results are shown as mean ± SEM from six independent experiments. ap < 0.05 vs. untreated cells, bp < 0.05 vs. Ang III-treated cells.
Role of AP-1 activation in Ang III-induced MCP-1 expression
To identify the intranuclear signaling pathway by which Ang III induces MCP-1 production, we pre-incubated cells with the c-Jun/AP-1 inhibitor curcumin and/or the NF-κB inhibitor PDTC for 30 minutes prior to addition of Ang III for the remaining 48 hours. MCP-1 protein levels in conditioned medium were measured by enzyme-linked immunosorbent assay. Pre-treatment with the AP-1 inhibitor significantly inhibited Ang III-induced MCP-1 production (p < 0.05), but the NF-κB inhibitor had no effect (Fig. 6).

Effects of the transcription factor inhibitors curcumin and pyrrolidine dithiocarbamate (PDTC) on angiotensin III (Ang III)-induced monocyte chemoattractant protein-1 (MCP-1) production. HK-2 cells were pre-incubated with curcumin or PDTC for 30 minutes and then incubated with Ang III (10−7 M) for 48 hours. MCP-1 protein levels were measured by enzyme-linked immunosorbent assay. Results are expressed as percentage increase over untreated cells. Results are shown as mean ± SEM from six independent experiments. ap < 0.05 vs. untreated cells, bp < 0.05 vs. Ang III-treated cells.
DISCUSSION
The aim of the present study was to examine whether Ang III is involved in monocyte recruitment by regulating the chemokine MCP-1. We demonstrated that Ang III increases MCP-1 production in HK-2 cells, partly through intracellular p38 and JNK MAPK signaling activity and subsequent AP-1 transcriptional activity.
Ang II is converted to Ang III by aminopeptidase A, which cleaves the NH2-terminal aspartate group and is present in glomerular and tubular cells [30]. The Ang II level is 1,000 times higher in the lumen of proximal tubular cells than in plasma and does not detectably decrease along the length of the proximal tubule [31]. Previous studies have shown that Ang III increases the renal inflammatory response in mesangial and mononuclear cells by a mechanism dependent on NF-κB and AP-1 [30-32]. However, it is unclear whether Ang III affects the monocyte-mediated inflammatory response in human renal proximal tubule cells such as HK-2 cells [32-34].
Our results suggest that Ang III upregulates MCP-1 and contributes to the accumulation of inflammatory cells such as monocytes in proximal tubule cells. Infiltration of monocytes into the kidney is thought to play a central role in progressive inflammatory renal disease [35,36]. Therefore, inhibiting Ang III may be important in managing inflammatory kidney disease.
Increased MCP-1 levels were inhibited by adding an AT1 receptor blocker (losartan), suggesting that the inflammatory response induced by Ang III is mediated by the AT1 receptor. Some effects of Ang II are also mediated by AT1, such as vasoconstriction, sodium reabsorption, sympathetic nerve stimulation, aldosterone secretion, and inhibition of renin synthesis [37,38]. Furthermore, Ang II binds to the AT2 receptor and contributes to the inhibitory effects mediated by AT1 [39]. In fact, which receptors Ang III acts on in renal cells is far from fully understood. Some studies show that Ang III activates NF-κB in cultured mesangial cells, mainly via AT2 receptors [18], and contributes to antinociception in rat brain cells via the AT1 receptor [40]. In addition, the baroreceptor reflex response at the rat nucleus tractus solitarii is modulated via both receptors [41]. It appears that Ang III has affinities to different receptor subtypes depending on the tissue and mechanism of action. Further studies are needed using selective AT1 and AT2 receptor antagonists or knockout animal models.
Aberrant or inappropriate MAPK function has been identified in many diseases, and MAPKs are ubiquitous elements in signaling pathways that control cellular functions [42]. In the present study, we evaluated the p38, JNK, and ERK MAPKs to determine which is involved in Ang III-stimulated MCP-1 production in HK-2 cells. Our results show that inhibiting p38 significantly decreases Ang III-induced MCP-1 production. Therefore, p38 may have a role in the intracellular signal pathway of Ang III-induced MCP-1 production in HK2 cells. Unlike ERK MAPKs, which are activated by growth factors, p38 MAPKs are responsive to stress stimuli, cytokines, and factors involved in cell differentiation and apoptosis, as are JNK MAPKs [43]. In our study, the total level of phosphorylated JNK appeared to increase suddenly 20 minutes after stimulation by Ang III, but the increase was not significant, likely due to small sample size. Additional study is needed to clarify whether JNK MAPKs also have a role in MCP-1 production.
The 5′-flanking region of the MCP-1 gene contains multiple AP-1 and NF-κB binding sites, suggesting a potential role for both intranuclear transcription factors in the regulation of MCP-1 expression [44]. Ang III-induced activation of NF-κB activity leading to increased MCP-1 expression in mesangial cells has been described previously [34]. However, this effect has not been demonstrated in proximal tubular epithelial cells. Therefore, we examined whether Ang III-induced activation of AP-1 or NF-κB is responsible for increased MCP-1 production in HK-2 cells.
Some studies have reported that the NF-κB and AP-1 transcription factors regulate MCP-1 expression in stimulus- and tissue-specific manners. In mesangial cells, interleukin (IL)-1β-induced MCP-1 expression is mediated by NF-κB [45,46], proteasome inhibitor-induced MCP-1 expression is mediated by the JNK/AP-1 pathway [47], and Ang III-induced MCP-1 expression is mediated by both NF-κB and AP-1 [34]. In particular, the role of AP-1 activation in the upregulation of MCP-1 expression has been described in various cells. Activation of AP-1 is required for inducing MCP-1 by IL-1β and shear stress in vascular endothelial cells [48,49]. However, no study has clearly determined whether Ang III-induced MCP-1 expression is mediated by AP-1 in proximal tubular epithelial cells such as HK-2 cells.
In our study, pre-treatment with the AP-1 inhibitor curcumin significantly inhibited Ang III-induced MCP-1 production. In fact, curcumin not only inhibits DNA binding of c-Jun/AP-1 transcription factors but also downregulates c-Jun gene transcription [50]. Therefore, we explored the role of c-Jun/AP-1 in Ang III-induced MCP-1 production using this inhibitor. When HK-2 cells were pre-incubated with curcumin, Ang III-induced MCP-1 production was inhibited. However, the NF-κB inhibitor PDTC did not block Ang III-induced MCP-1 production. These results indicate that AP-1 activation is involved in Ang III-induced MCP-1 expression. c-Jun contains a docking site for JNKs, which phosphorylates serines 63 and 73, thereby enhancing its transcriptional activity and stability [51]. Activation of JNK or p38 MAPK results in greatly enhanced c-Jun transcriptional activity and induction of AP-1 target genes [51,52].
In conclusion, our results suggest that Ang III induces MCP-1 expression in HK-2 cells by increasing JNK and p38 activities and subsequent AP-1 activity. Our results also provide a rationale to investigate inhibition of Ang III as a potential new therapeutic target in inflammatory renal disease.
KEY MESSAGE
1. Angiotensin III (Ang III) increases monocyte chemoattractant protein-1 synthesis in HK-2 cells by stimulating p38 and c-Jun N-terminal kinases mitogen-activated protein kinase activities and subsequent activating protein-1 activity.
2. Our results provide a rationale to investigate the inhibition of Ang III as a potential new therapeutic target in inflammatory renal disease.
Notes
No potential conflict of interest relevant to this article was reported.