Evaluation of the effect of D-002, a mixture of beeswax alcohols, on osteoarthritis symptoms
Article information
Abstract
Background/Aims
Nonsteroidal anti-inflammatory drugs relieve osteoarthritis (OA) symptoms but cause adverse effects. D-002, a mixture of beeswax alcohols, is effective against experimental OA. A pilot study found that D-002 (50 mg/day) for 8 weeks improves OA symptoms. The aim of this study was to investigate the effects of D-002 (50 to 100 mg/day) administered for 6 weeks on OA symptoms.
Methods
Patients with OA symptoms were double-blindly randomized to D-002 (50 mg) or placebo for 6 weeks. Symptoms were assessed by the Western Ontario and McMaster Individual Osteoarthritis Index (WOMAC) and the visual analog scale (VAS) scores. Patients without symptom improvement at week 3 were titrated to two daily tablets. The primary outcome was the total WOMAC score. WOMAC pain, joint stiffness and physical function scores, VAS score, and use of rescue medications were secondary outcomes.
Results
All randomized patients (n = 60) completed the study, and 23 experienced dose titration (two in the D-002 and 21 in the placebo groups). At study completion, D-002 reduced total WOMAC (65.4%), pain (54.9%), joint stiffness (76.8%), and physical function (66.9%) WOMAC scores, and the VAS score (46.8%) versus placebo. These reductions were significant beginning in the second week, and became enhanced during the trial. The use of rescue medication by the D-002 (6/30) group was lower than that in the placebo (17/30) group. The treatment was well tolerated. Seven patients (two in the D-002 and five in the placebo group) reported adverse events.
Conclusions
These results indicate that D-002 (50 to 100 mg/day) for 6 weeks ameliorated arthritic symptoms and was well tolerated.
INTRODUCTION
Osteoarthritis (OA) is a multifactorial painful and disabling degenerative joint disease involving the cartilage and many surrounding tissues and is one of the most common musculoskeletal disorders affecting hundreds of millions worldwide, but mainly the elderly [1-3].
Although nonpharmacological interventions are the mainstream treatment for OA management [4], current guidelines support the use of analgesics such as paracetamol, and nonsteroidal anti-inflammatory drugs (NSAIDs) to provide symptom relief in patients with OA, despite that they do not solve the underlying pathological process. Optimal treatment combines both nonpharmacological and pharmacological modalities [5,6]. Nevertheless, a search for safer alternatives is justified due to gastrointestinal adverse effects (AE) of nonselective NSAIDs, the cardiovascular AE of cyclooxygenase 2 (COX-2) inhibitors, and the hepatotoxicity of paracetamol [7-10].
D-002 is a mixture of six high-molecular-weight aliphatic alcohols (C24, C26, C28, C30, C32, and C34) purified from beeswax [11], which inhibits both cyclooxygenase (COX) and 5-lipooxygenase (5-LOX) enzyme activities in vitro (Perez et al. in press); thus, acting as a dual anti-inflammatory substance, which results in a safer gastrointestinal profile compared to that of NSAIDs [12]. Consequently, D-002 is effective in experimental models of acute and chronic inflammation [13,14] and experimental OA [15]. In addition, preliminary results of a pilot study indicated that D-002 (50 mg/day) administered for 8 weeks led to improvement in OA symptoms [16].
Thus, the present study was conducted to investigate the effects of D-002 (50 to 100 mg/day) administered for 6 weeks on OA symptoms.
METHODS
Study design
This randomized, double-blind, placebo-controlled study was approved by the Institutional Ethics Committee of the Surgical Research Centre (Havana, Cuba) and was conducted in accordance with the ethical standards established in the Declaration of Helsinki.
Patients were provided oral and written explanations of the nature of the trial and the study treatment and gave their informed written consent at enrolment.
Eligible patients were randomized to D-002 (50 mg) or placebo tablets, which were taken once daily with breakfast for 6 weeks. Thereafter, subjects attended a visit each week. Physical examinations and a symptom assessment were conducted at each visit. Treatment compliance, control of rescue analgesic consumption, and AE were controlled weekly, whereas laboratory examinations were performed at baseline and after 3 and 6 weeks of treatment.
Study participants
Ambulatory females and males 20 to 80 years of age with a prior diagnosis of OA of the knee, hip, or fingers and supported by clinical and radiological criteria were enrolled in the trial. Participants were required to have a diagnosis of functional class I, II, or III OA (mild to moderate) in accordance with the American College of Rheumatology Criteria [17,18] and a Western Ontario and McMaster Individual Osteoarthritis Index (WOMAC) score ≥ 30 [19-22].
Exclusion criteria were suffering from other forms of arthritis, arthroscopy within the past year, intra-articular injection of steroids within the past 3 months, uncontrolled hypertension (diastolic pressure ≥ 120 mmHg) or diabetes (fasting glucose > 7 mmol/L), active liver or renal disease, malignancies, any other serious illnesses, hospitalization during the 6 months prior to the study or the following laboratory abnormalities: alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) > 45 U/L, and creatinine > 130 µmol/L. In addition, pregnant or nursing females, and those not taking adequate contraceptive measures were excluded from the trial.
Predefined premature discontinuations included unwillingness to follow-up, an AE supporting such a decision, and protocol violations (failure to take treatment ≥ 5 days).
Treatment
The study tablets were produced under Licensees and Good Manufacturing Practices conditions and came directly from the manufacturers (Plants of Natural Products, Laboratorios MedSol, Havana, Cuba). D-002 content was assessed by gas chromatography [23]. The placebo had a similar composition as the D-002 tablets, except the active ingredient was replaced with lactose. Otherwise the placebo and D-002 tablets were indistinguishable. Treatments were packaged in identical PVC-aluminum sealed blister packs.
At visit 2, the coded and packaged study tablets (D-002 50 mg or placebo) were given to the study subjects. Tablets were taken once daily with breakfast for 6 weeks, but patients without symptom improvement by week 3 were titrated to two tablets daily. Then, D-002-treated patients received a dose of 100 mg/day.
The randomization code was computer-generated with a fixed, not stratified randomization method, using balanced blocks and an allocation ratio of 1:1. The starting dose of D-002 (50 mg/day) selected was the same used in the 8-week pilot trial in subjects with OA symptoms [16].
The entire code was kept confidential at the generating location. Sealed individual envelopes with codes of each subject were kept at the generating place and at the site of the Principal Investigator and were opened prematurely in case of a serious adverse event (SAE) occurred, but such a situation did not occur in the trial.
Treatment compliance was controlled by counting the remaining tablets and interviewing participants. At trial completion, unused tablets were recovered. Compliance was considered good if the subjects consumed at least 85% of the tablets scheduled from the previous visit.
Subjects were not allowed to consume NSAIDs, steroids, cartilage or calcium supplements, or any other agent that may have affected the study outcome, except rescue medications to treat persistent pain: acetaminophen (maximum 2 g/day) or metamizole (maximum 600 mg/day). All patients were instructed to keep a diary of their consumption of rescue medication and report them at their next scheduled visit. The number of consumed rescue medication tablets was recorded at each visit.
Outcome measures
The primary endpoint was to obtain a significant reduction in the total WOMAC index of not less than 30% compared to that of the placebo group (Table 1) [19-22]. The WOMAC questionnaire is used widely in clinical trials to evaluate the effect of investigational products on OA, provides a validated assessment of the patient's functional capacity, specifically joint pain, stiffness, and functional impairment [19-22].

Modified Western Ontario and McMaster Individual Osteoarthritis Index questionnaire
At each visit, subjects completed the WOMAC questionnaire, which consists of three sections, one that assesses pain intensity (five questions), other joint stiffness (two questions), and the third that assesses physical function (17 questions). Individual responses were scored on the following scale: 0 (none), 1 (slight), 2 (moderate), 3 (severe), and 4 (extreme). The total score range was 0 (the best) to 96 (the worst).
Reductions in pain, stiffness, and physical function as assessed by the WOMAC score and a visual analog scale (VAS) for pain were secondary efficacy variables [24,25]. The treatment should significantly decrease scores compared to placebo to be considered effective. Patients completed the WOMAC and VAS questionnaires in the physician's office before their examination to avoid bias. The VAS score used a 100-mm linear measure of pain status with 0 representing no pain and 100 the worst imaginable pain. Patients marked the relevant amount of pain they were experiencing on a linear scale.
A decrease in the use of rescue medications versus placebo was another secondary outcome. The amount of rescue medication was assessed in terms of total use at the conclusion of the study.
Finally, the patient's perception of treatment efficacy in terms of symptom relief was a collateral outcome. Responses were classified as follows: very good (complete symptom relief), good (partial, but relevant, symptom relief), fair (modest improvement), or nil (symptoms unchanged). In this case, the goal was that the frequency of responses classifying efficacy as very good or good in the treatment group should be ≥ 70% and significantly greater than that in the placebo group.
Safety and tolerability assessment
Safety indicators included vital signs (body weight, pulse rate, blood diastolic and systolic pressure), and blood indicators (erythrocyte sedimentation rate, ALT, AST, serum fasting glucose, and creatinine). Blood biochemical safety indicators were assessed with enzymatic methods using reagent kits (Roche, Basel, Switzerland) and performed on a Hitachi 709 Autoanalyser (Tokyo, Japan). Erythrocyte sedimentation rate was assessed by a conventional method at the clinical laboratory of the Surgical and Medical Research Centre (Havana, Cuba). Controls for precision and accuracy of the methods were performed.
We considered all AEs undesirable events that occurred during the study. Subjects were queried by investigators regarding the occurrence of AEs between study visits. AEs were recorded on the case record form, including the characteristics, dates of onset and disappearance, treatments adopted, and responses achieved. The severity of the AE was classified as mild, moderate or serious; mild, were those easily tolerated AEs that did not require suspension of study medication and/or specific treatment; moderate, those that caused discomfort enough that therapy and/or specific treatment was stopped; and SAE, those disabling events that leaded to hospitalization and/or death. AEs that occurred within 30 days of consuming the last study dose, monitored by direct contact with the subjects, were included in this analysis. The causal relationships between AEs and the treatments were classified using the Naranjo algorithm [26].
Statistical analysis
Data were analyzed using an intention-to-treat approach. Thus, data of all randomized subjects were included in all analyses. The sample size estimate assumed a difference of ≥ 30% between the reduction in WOMAC total score from baseline in the D-002 and placebo groups at study completion. Then, 30 subjects per treatment arm were sufficient to detect such a difference with 80% power and α = 0.05. Assuming a permissible dropout rate of 10%, 66 subjects were enrolled.
The Mann-Whitney U-test for comparisons between groups of continuous data and the Wilcoxon test for paired samples with Bonferroni adjustment were used for multiple comparisons, as appropriate. Categorical variables were compared with Fisher exact probability test. All statistical tests were two-tailed with α = 0.05, and p < 0.05 was considered to indicate significance. Statistics software for Windows and MS Excel (Microsoft Co., Redmond, WA, USA) were used for the analysis.
RESULTS
Baseline characteristics
Sixty-seven subjects were recruited. Of them, 60 were eligible for randomization. The remaining seven subjects did not pass to the active treatment step because of fasting glucose > 7 mmol/L (three subjects), and WOMAC scores < 30 (four cases). All randomized patients (100%) completed the study.
The baseline characteristics of the groups were well matched, so that subject randomization was effective (Table 2) [27-29]. Gender was predominantly female (37 females and 23 males). Forty-two subjects (70%) were above normal weight (29 were overweight, 13 were obese). In addition, the frequencies of hypertension (65%) and hypercholesterolemia (36.7%) among study subjects were relatively high due to lifestyle factors such as being sedentary (40%) and smoking (26.7%). The frequency of consumption of concomitant medication was high among randomized subjects (51/60, 85%).

Baseline demographic and clinical characteristics of the study population
Efficacy analysis
Treatment compliance was very good and similar in both groups. Twenty-three subjects were titrated to two daily tablets (2/30 in D-002, 21/30 in the placebo group; p < 0.0001). Only two D-002 patients received 100 mg/day during the last 3 weeks of the study.
At baseline, the total WOMAC scores (mean ± standard deviation [SD]) were 38.8 ± 8.2 (D-002 group) and 38.6 ± 8.4 (placebo) and comparable between the groups (Table 3). No significant changes were seen at week 1. After 1 week of treatment, the D-002 group had a decreased (p < 0.001) total WOMAC score by 12.9% compared to a slight decrease (3.1%) in the placebo group (difference of 9.8% vs. placebo). The treatment effect did not wear off but became enhanced during the treatment period. At study completion, the total WOMAC score in the placebo group (34.1 ± 8.5) was similar to that observed at baseline, whereas it decreased significantly (p < 0.00001 vs. baseline and placebo) to 8.9 ± 6.2 in the D-002-treated group (77.1% decrease vs. baseline, 65.4% vs. placebo).
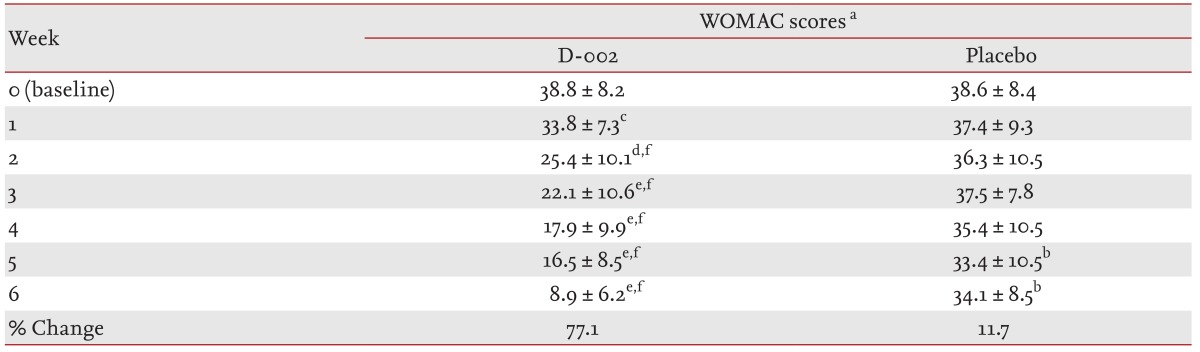
Changes in the total Western Ontario and McMaster Individual Osteoarthritis Index scores
The mean ± SD baseline WOMAC pain scores were 8.7 ± 2.4 (D-002) and 9.2 ± 2.9 (placebo) (Table 4). After 1 week of treatment, the pain score had decreased significantly in the D-002 group (20.7% decrease vs. baseline, p < 0.001; 17.4% vs. placebo, p < 0.01). The effect was more pronounced during the trial, so that at the end of the trial WOMAC pain score in the D-002 group was significantly lower (p < 0.00001) by 69.0% (vs. baseline) and 54.9% (net reduction vs. placebo).

Changes in pain, stiffness, and physical function on the Western Ontario and McMaster Individual Osteoarthritis Index scores by treatment group
At randomization, the mean stiffness score was 2.9 ± 0.9 in the D-002 group and 3.1 ± 1.0 in the placebo group. At week 1, the score decreased significantly with D-002 (27.6% vs. baseline, p < 0.001; 17.9% vs. placebo, p < 0.05). At study completion, reduced values for stiffness in the D-002 group (p < 0.00001 vs. baseline and placebo) were 89.7% and 76.8% compared to baseline and the placebo, respectively.
The sequential changes in WOMAC physical function scores were similar to those referred above for the other WOMAC scores. The mean baseline values of the D-002 (27.1 ± 5.8) and placebo (26.4 ± 6.4) groups were similar; scores at 2, 4, and 6 weeks of treatment were significantly lower (p < 0.001 for week 2, p < 0.00001 for weeks 4 and 6) than those in the placebo group. The lower score in the D-002 group was accentuated over the 6 week period, so that it was 77.9% and 66.9% at trial completion compared to baseline and placebo, respectively.
Table 5 summarizes the mean VAS scores. Both groups exhibited similar values at randomization; 63.3 ± 13.9 in the D-002 group and 63.5 ± 19.0 in the placebo group. After 1 week of D-002 treatment, the VAS score decreased significantly compared to that at baseline (p < 0.001). This decreased at the end of the trial was 60.2% and 46.8% of baseline (p < 0.00001) and placebo (p < 0.0001).
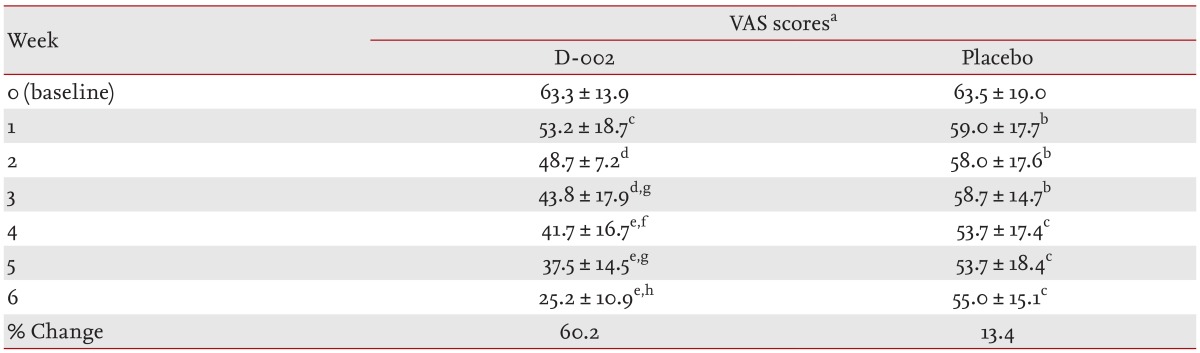
Changes in visual analog scale scores by treatment group
Twenty-eight of thirty D-002-treated subjects (93.3%) had very good (6/30) or good (22/30) efficacy, whereas only 9/30 (30.0%) subjects from the placebo group had good efficacy (p < 0.0001 vs. placebo). In contrast, while 1/30 (70%) placebo subjects felt that efficacy was fair (14/30) or nil (7/30, 23.3%), only two subjects in the D-002 group (6.7%) stated that their efficacy was fair (p < 0.001 vs. placebo).
Twenty-three participants (six in the D-002 and 17 in the placebo) consumed acetaminophen or metamizole during the trial. Consumption of rescue medications in the D-002 group (6/30) was less frequent (p < 0.01) than in the placebo group (17/30).
Safety and tolerability
The treatment was well tolerated. There were no withdrawals from the study. Seven patients (two in the D-002 and five in the placebo) reported an AE during the study (Table 6).
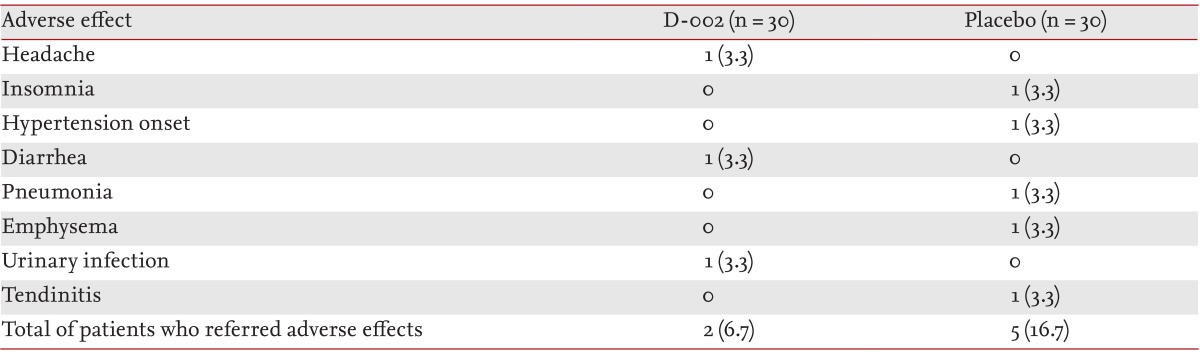
Adverse effects during the study
Vital signs and blood parameters did not change significantly during the study, and individual values remained within normal ranges (data not shown).
DISCUSSION
Pain and impaired mobility are common OA symptoms that affect the life of sufferers, which currently includes millions of people worldwide [1,2].
We found that patients with OA symptoms treated with D-002 (50 to 100 mg/day) for 6 weeks showed significantly improved pain, stiffness, physical functioning, and total WOMAC and VAS scores for pain compared to those in the placebo-controlled group, and that such benefits were well perceived by the study patients. The effects, mostly observed after 1 to 2 weeks of treatment, were enhanced throughout the study.
Both groups were homogeneous at baseline, indicating that the randomization was adequate and that results are attributable to the D-002 treatment, rather than to initial differences between the groups. The mean age of the study subjects (67 years) was consistent with that expected for this disease, which manifests in mainly elderly people [1-3]. Females (61.7%) outnumbered males (38.3%), and a higher prevalence of OA was observed in postmenopausal females (31 of 37 randomized females [83.8%]) [30,31]. The high frequencies (≥ 20%) of overweight plus obesity (70%), hypertension (65%), sedentary lifestyle (40.0%), hypercholesterolemia (36.7%), and smoking (26.7%) among study subjects reflects not only the occurrence of concomitant coronary risk factors, common in Cuban subjects of this age [32], but agrees with reports of comorbid conditions in middle-aged and older subjects with OA [33].
D-002 treatment produced significant reductions in total (primary outcome) and subset (secondary outcomes) WOMAC scores beginning at week 2, and were enhanced thereafter, so that at study completion the total WOMAC score had decreased by 77.1% versus baseline, 65.4% versus placebo. Subjects experienced less pain and stiffness, and the physical WOMAC scores decreased by 54.9%, 76.8%, and 66.9% versus placebo, respectively.
The significant decrease in the VAS score exhibited a similar pattern. The score was significantly higher beginning after the first week of treatment and accentuated progressively over the trial, with final decreases of 60.2% and 46.8% compared to baseline and placebo, respectively. Despite the differences in WOMAC and VAS scales, the decreased pain detected by these tools in the D-002 (decreases vs. placebo of 54.9% and 46.8% in the WOMAC pain and VAS scores, respectively) and placebo groups (reductions vs. baseline of 14.1% and 13.4% with WOMAC pain and VAS scores, respectively) were comparable.
Modest and clinically nonmeaningful (≤ 15%) decreases in total pain, stiffness, physical function and total WOMAC scores and VAS score were seen in the placebo group at the end of the study. However, this mild improvement in the placebo should not be surprising, as it may occur in any efficacy measurement based on a subjective assessment and, particularly, in placebo-controlled studies in subjects with OA [34]. Possible explanations for this include that participants were advised to remain physically active during the treatment, such as walking 30 minutes every day; and that 17/30 of placebo subjects (56.7%) used rescue medications during the trial. However, this small placebo effect does not limit the efficacy of D-002 for ameliorating OA symptoms seen here. The significance and magnitude of the decreases in the scores following D-002 administration and the reduced use of rescue medication is clear evidence that the efficacy results were attributable to D-002. Twenty-eight subjects (93.3%) perceived that treatment efficacy was very good (6) or good (22), whereas only nine (30.0%) in the placebo group had good efficacy. The significant difference in the dose titration between the groups (two in the D-002 and 21 in the placebo) indirectly supports the treatment efficacy result.
Although these data are consistent with those of a previous 8-week pilot study [16], the effects of D-002 on WOMAC scores seen here were greater than those reported previously. No marked differences in the baseline characteristics of the study participants were observed, with the exception of a predominance of elderly people and males, which explains the higher efficacy of D-002 in this shorter-duration study. As only two D-002-treated patients were titrated to 100 mg/day during the last 3 weeks, dose titration did not seem to explain the difference. Although the lower score in the placebo group was similar to that found previously (11.7% vs. 11.9%), the observation that all subjects completed the study and that treatment compliance was very good reflects an excellent patient/physician rapport. This could have contributed to the subjects performing the recommended daily 30 minutes of walking, which may have resulted in improved results in conjunction with the treatment.
The detailed mechanisms by which D-002 alleviates OA symptoms are unknown and their elucidation is beyond the scope of this study. Nevertheless, the mechanisms are probably associated with the dual anti-inflammatory effect of D-002, demonstrated experimentally [13,14], as D-002 reduces COX and 5-LOX activities in vitro and leukotriene B4 content in pleural exudates of rats with carrageenan-pleurisy [13].
The present results are in line with those from studies of supplements with anti-inflammatory effects and a good gastrointestinal safety profile. These supplements have shown benefits in experimental arthritis and in patients with OA, similar to the case of dietary n-3 polyunsaturated fatty acids, which inhibit COX activity and when given together with glucosamine sulfate are more effective compared to glucosamine alone in patients with OA [35-37].
Other complementary treatments seem to be useful for managing OA symptoms. Glucosamine sulfate has a carryover effect similar to disease-modifying agents, and long-term treatment with this agent may lower dependence on NSAIDs and delay OA progression [36]. A meta-analysis suggested that a specific preparation of chondroitin sulfate (Structum, Laboratoires Pierre Fabre, Castres, France) (1 g/day) given for 3 to 6 months is effective in terms of reducing pain in patients with symptomatic knee OA, supporting the use of chondroitin sulfate to manage symptomatic knee OA [37].
Oral treatment with D-002 is gastroprotective, in contrast to NSAID-related gastrotoxicity [38-42]. The mechanism whereby D-002 protects the gastric mucosa involves increased secretion and improved composition of the gastric mucus [39,40], and a reduction in lipid peroxidation, generation of hydroxyl radicals, and protein oxidation in the gastric mucosa [41,42]. The antioxidant effects of D-002 have been demonstrated in experimental and clinical studies [43-46]; this is in marked contrast to the NSAID-related gastrointestinal safety profile.
D-002 treatment was safe and well tolerated, which is consistent with previous clinical studies [16,44-46], and the absence of gastrointestinal AEs matched the gastroprotective effects of D-002.
The current concern over the increased cardiovascular and stroke risk associated with COX-2 inhibitors, and the gastrointestinal and renal complications produced by nonselective NSAIDs support the need for new therapies, including complementary medicine for OA management. In such a scenario, the present results suggest that D-002 treatment (50 to 100 mg/day), which is devoid of gastrotoxic effects, could be useful for management of OA symptoms. However, these results are preliminary, and the efficacy of D-002 in patients with OA should be investigated extensively in further studies.
The results indicate that D-002 (50 to 100 mg/day) for 6 weeks ameliorated arthritic symptoms and was well tolerated. D-002 could be beneficial for managing OA symptoms but extensive clinical research is necessary to verify our findings.
KEY MESSAGE
Oral administration of D-002 (50 to 100 mg/day) for 6 weeks may ameliorate arthritic symptoms meanwhile improve clinical evolution in patients with osteoarthritis (OA).
D-002 seems to be safe and well tolerated in patients with OA.
Notes
No potential conflict of interest relevant to this article was reported.