Three Cases of Congenital Hepatic Fibrosis with Caroli’s Disease in Three Siblings
Article information
Abstract
Congenital hepatic fibrosis is a relatively rare disease of children and young adults characterized by hard hepatomegaly, portal hypertension with relative preservation of liver function and underlying architecture, and frequent renal involvement.
We experienced 3 cases of congenital hepatic fibrosis with Caroli’s disease in 3 siblings, whose clinical manifestations were diverse, such as repeated cholangitis, variceal hemorrhage, or intrahepatic stones. All of them had multiple renal cysts, so we supposed that the clinical entities of these patients were in the spectrum of fibropolycystic disease of the liver and kidney.
INTRODUCTION
Congenital hepatic fibrosis is an inherited, congenital malformation characterized by large, fibrotic portal spaces that contain multiple bile ductules, the main consequence of which is portal hypertension1).
Congenital hepatic fibrosis frequently does not exist as a single entity but as members of a family. The members are found in various combinations. They consist of polycystic liver disease, congenital hepatic fibrosis, Caroli’s disease, choledochal cysts, and renal cysts2–4).
We report 3 cases of congenital hepatic fibrosis with Caroli’s disease in 3 siblings, who also had multiple renal cysts. This report also reviews the pathogenesis of congenital hepatic fibrosis and the possible relationship with Caroli’s disease and associated anomaly.
CASE REPORT
1. Case 1
This 33-year-old man was admitted via the emergency room due to recurrent fever and right upper quadrant abdominal pain. Since his childhood he suffered from easy fatigue and was informed that he had had a palpable enlarged spleen from birth. His younger brother had been previously diagnosed at private clinic 3 years ago with congenital spherocytosis due to splenomegaly, pancytopenia, and increased peripheral ovalocytes. Therefore, this 33-year-old patient was also evaluated and received splenectomy several months later under the impression of congenital spherocytosis (At that time, his RBC fragility test was negative, but peripheral ovalocytes was 26%). According to operative findings at the clinic, his liver was grossly nodular and enlarged, so a liver biopsy was done. Following the biopsy, he was diagnosed at the private clinic as having liver cirrhosis.
Seven months before entering our hospital, diarrhea with fever and chills developed. At that time, he was diagnosed with amebic colitis and treated with antibiotics. Four months before admission to our hospital, right upper quadrant abdominal pain with fever developed, so he was checked and managed as a cholecystitis patient. However, the fever and abdominal pain continued to fluctuate in severity thereafter. Two days prior to his admission to our hospital, severe abdominal pain and fever were aggravated.
On admission to our hospital, the patient’s temperature was 39.3°C, the pulse 92, and the respiration 24. The blood pressure was 110/70 mmHg. On examination, the patient was relatively well-nourished but appeared acutely ill. No rash, lymphadenopathy, or jaundice was evident. The liver was 3 finger-breadth’s palpable on the epigastrium, which was hard and tender. The kidney was not palpable. We reviewed the microscopic findings of the obtained previous liver biopsy specimen, which had been done 3 years ago. Microscopic finding showed preserved normal hepatic cell cords with diffuse portal and periportal fibrosis surrounded by proliferated bile ductules and bizarre-shaped ectatic bile ducts (Fig. 1). Abdominal computed tomography findings revealed Caroli’s disease and hepatomegaly accompanied by multiple renal cysts (Fig. 2). We diagnosed him with congenital hepatic fibrosis and Caroli’s disease, complicated by cholangitis. As a result, due to suspicion of hereditary causes, we reevaluated and tested his family members, including the Case 2 and Case 3 patients. The laboratory and imaging findings are summarized in Table 1.
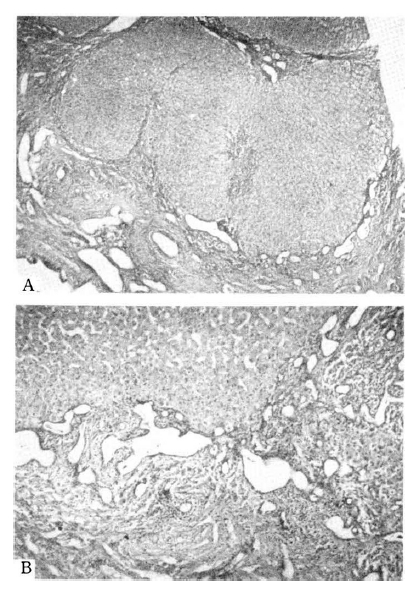
The Liver Biopsy Finding in Case 1.
A. Proliferation and dilatation of the bile ducts and diffuse portal fibrosis are shown. H & E stain, magnification × 100. B. There are relatively well preserved hepatic cell cords surrounded by fibrosis and bizarre-shaped ectatic bile ductules. H & E stain, magnification × 250.
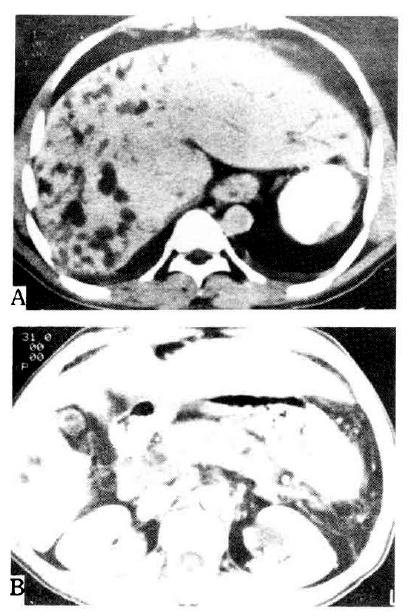
Abdominal computed tomography in case 1. A. Hepatomegaly with dilatation of the intrahepatic bile duct is demonstrated. The spleen is not seen due to previous splenectomy. B. Multiple cysts are noted in both kidneys.
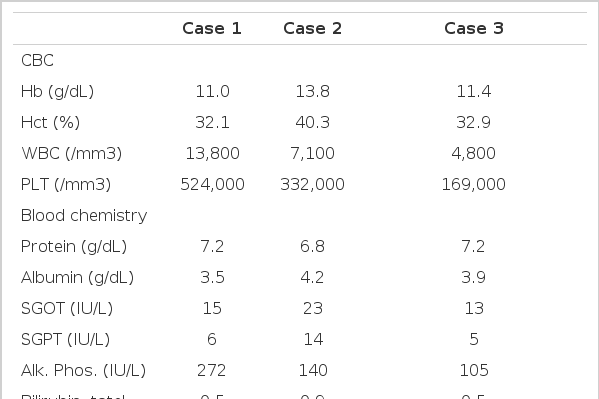
Laboratory and Imaging Findings
2. Case 2
This 27-year-old male is the younger brother of the Case 1 patient. He also had a history of splenomegaly from birth. In infancy he had suffered from poliomyelitis from which residual poliomyelitis and scoliosis remained. Three years before admission to our hospital, he had visited a private clinic for correction of scoliosis and was diagnosed with congenital spherocytosis due to splenomegaly, pancytopenia, and increased peripheral ovalocytes. So he received splenectomy at that hospital. According to the operative findings, his liver was grossly nodular and enlarged, so he was managed as one with liver cirrhosis. Several months later after splenectomy, massive hematemesis developed and esophageal varix was detected.
During his admission period in our hospital, massive hematemesis recurred, so he was treated by endoscopic sclerotherapy. Gastroscopic findings showed severe esophageal varix with red color sign. Abdominal computed tomography revealed Caroli’s disease, hepatomegaly with increased collateral circulation, and multiple renal cysts (Fig. 3). Portography showed multiple periportal collaterals and loss of visualization of the main portal vein. The laboratory and imaging findings are summarized in Table 1.
Abdominal computed tomography in Case 2. A. Hepatomegalv with dilatation of the intrahepatic bile duct is noted. The spleen and gallbladder are not seen due to previous splenectomy and cholecystectomy. B. Multiple renal cysts and marked scoliosis are noted.
3. Case 3
This 35-year-old male is the elder brother of the Case 1 patient. He also had a history of splenomegaly from birth. At age 8 he was found to have bladder stones, so an operation for stone removal was performed. After that he had been well, but during his military service, about 10 years before admission to our hospital, sudden severe abdominal pain developed so he received exploratory laparotomy. According to the operative findings, his liver was also grossly nodular and enlarged, so he was diagnosed with liver cirrhosis but no specific treatment was given. After that he has had recurrent episodes of right upper quadrant abdominal pain and fever.
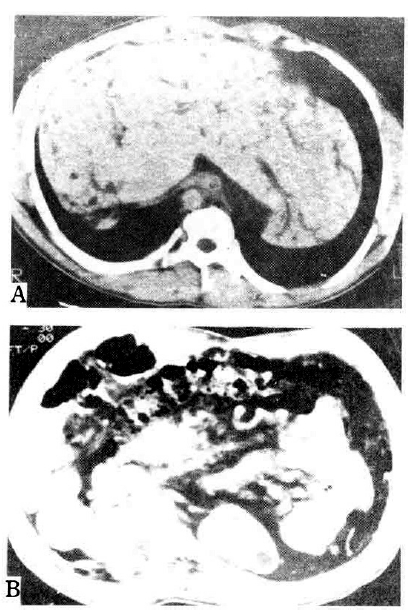
In our hospital, endoscopic retrograde cholangiopancreatography showed Caroli’s disease with multiple intrahepatic duct stones, while abdominal computed tomography revealed hepatosplenomegaly and multiple cysts of the liver and kidney in addition to the finding of Caroli’s disease (Fig. 4). The laboratory and imaging findings are summarized in Table 1.
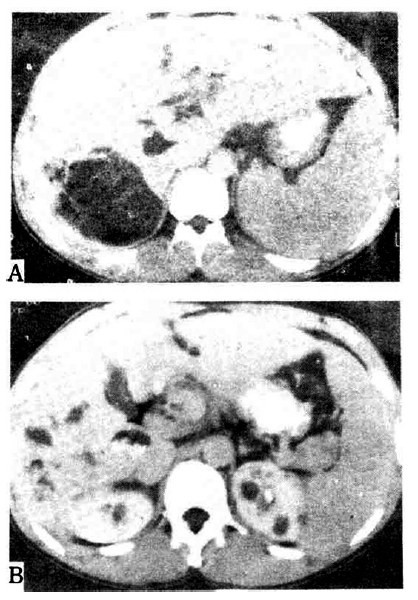
Abbominal computed tomography in Case 3. A. Hepatosplenomegaly and dilated intrahepatic bile duct with stones are demonstrated. There are also multiple hepatic cysts. B. Multiple cysts are noted in both kidneys.
DISCUSSION
The primary disorder of congenital hepatic fibrosis is likely to be bile ductular proliferation, with fibrosis being secondarily induced by the multiple bile ductules1). Bile duct proliferation is thought to be a form of biliary dysgenesis arising from arrested biliary development3,5,6). The proliferated and ectatic bile ductal changes might be caused by a combination of an uneven and disproportionate overgrowth of biliary epithelia and their supporting connective tissue1). A similar disorder affecting the epithelium of the large bile ducts might account for Caroli’s disease associated with congenital hepatic fibrosis7). A similar mechanism might explain the dilatation of the renal collecting tubules. That is, an identical insult at different loci may result in choledochal cyst, dilatations of the major intrahepatic bile ducts (Caroli’s disease), congenital hepatic fibrosis, polycystic liver and kidney disease, or a combination of these anomalies1–3,5,8). In our cases, all 3 patients had congenital hepatic fibrosis, Caroli’s disease, and multiple renal cysts. Case 3 also had multiple cysts in the liver. This supports the hypothesis that congenital hepatic fibrosis, Caroli’s disease, choledochal cyst, land multiple cystic lesions of the liver and kidney may be closely related entities representing different levels of involvement of the same basic congenital defect5).
In congenital hepatic fibrosis, diverse clinical manifestations, such as portal hypertension, renal failure, cholangitis or mixed form, can be shown with or without associated anomaly4,8–10).
Portal hypertension does not develop in Caroli’s disease. In contrast to Caroli’s disease, portal hypertension may develop relatively early in congenital hepatic fibrosis and may progress to a fatal termination by bleeding esophageal varices8–10). In our cases, all 3 patients had portal hypertension which was evident by hypersplenism, esophageal varices, or increased periportal collaterals. But their liver function was well preserved (Table 1). Portal hypertension in congenital hepatic fibrosis is presinusoidal, and the liver function is usually preserved9). The long-term outcome for patients with the portal hypertension variety is fairly good11). The patient’s death was due to massive variceal bleeding but not to liver failure12).
The clinical manifestations of the cholangitic variety depends mainly on the location of the associated anomaly along the biliary tree13). Dilatations of the major extrahepatic (choledochal cyst) or intraheptic (Caroli’s disease) bile duct are associated with bile stasis and progressive liver disease, whereas patchy lesions involving only the terminal interlobular bile ducts (congenital hepatic fibrosis) or noncommunicating parenchymal cysts (polycystic liver disease) produce little disturbance of bile flow. Bile stasis and infection complicate sepsis6) and intrahepatic duct stones14). The cholangitic variety has a poor prognosis, and recurrences may lead to death15,16). In our patients, Case 1 and Case 3 had suffered from recurrent episodes of right upper quadrant abdominal pain and fever, suggested to be a manifestation of Caroli’s disease complicated by cholangitis.
Congenital hepatic fibrosis may present in the perinatal or neonatal period, when the emphasis is on the associated renal disease9,17,18).
The younger the patient, the more renal tubules are involved, so that the perinatal patient dies in renal failure. In the childhood and adult types, only a few tubules are involved, and the presentation is of hepatomegaly or portal hypertension19,20). In general, the renal lesion is more important in the newborn and infant, whereas the hepatic lesion dominates the picture in older children and adolescents21). In our cases, all 3 patients had multiple renal cortical cysts. Renal function was mildly impaired in Case 1 and Case 2. Mild proteinuria and somewhat decreased creatinine clearance were observed in Case 1 and Case 2, but intravenous pyelography was normal in all 3 patients (Table 1).
Congenital hepatic fibrosis is characterized pathologically by diffuse portal and perilobular fibrosis and multiple irregular bizarre-shaped bile ductules in the area of fibrosis1,20). Apparent progression to perilobular fibrosis with parenchymal nodularity grossly resembled cirrhosis6). But in contrast to liver cirrhosis, hepatic lobular architecture is normal, and liver function is well preserved as shown is Case 1 (Fig. 1, Table 1). Although liver biopsy was done in Case 1 only, splenomegaly from birth, previous operative findings of grossly nodular liver surface and hepatomegaly, preserved liver function in spite of portal hypertension in 3 siblings, and typical liver biopsy findings in Case 1 are believed to be sufficient conditions to confirm a diagnosis of congenital hepatic fibrosis12) in our 3 cases.
Our cases showed diverse clinical manifestations such as repeated cholangitis in Case 1, massive variceal hemorrhage in Case 2, and intrahepatic duct stones associated with cholangitis in Case 3. The Case 1 and Case 2 patients had received a splenectomy at a private clinic under an incorrect impression of congenital spherocytosis, and then they were managed as liver cirrhosis patients. We suspect these patients showed increased peripheral spherocytes due to hypersplenism from portal hypertension and were misdiagnosed as liver cirrhosis due to an apparent nodular enlarged liver. An autohemolysis test, which was done in our hospital, was negative in these patients. The Case 1 and Case 2 patients had been relatively well until they received the splenectomy. Since that time, repeated cholangitis developed in Case 1 and recurrent variceal hemorrhage in Case 2.
Congenital hepatic fibrosis is a developmental abnormality that may appear either sporadically or in several members of a family, whose parents are normal9). It probably is an inherited autosomal recessive disease that affects both sexes3,4). In the pedigree of our presented cases, the exact inherited pattern could not be shown due to multiple artificial abortion. But 3 males and 1 female including the presented cases are evident to be affected. The affected female, who was also known to have had a splenomegaly, died at age 5 (Fig. 5).
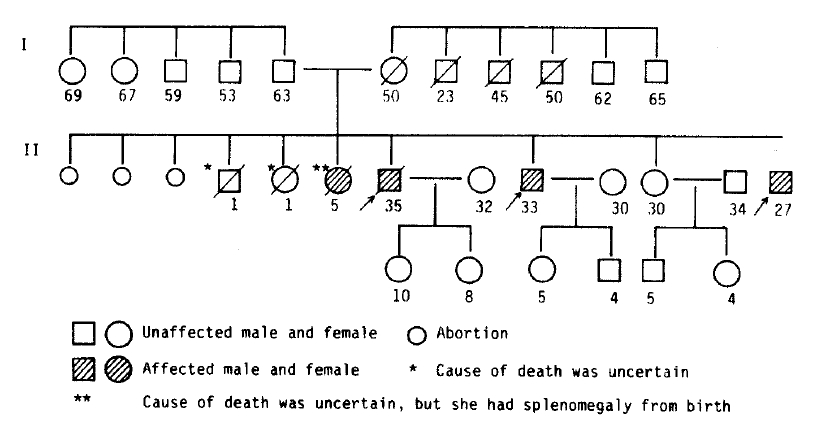
The pedigree of presented cases.
The arrows indicate the presented cases, and the numeral letter represents the age.
After discharge, the Case 3 patient has been relatively well, but the Case 1 and Case 2 patients are suffering from episodic high fever and chills, which are poorly controlled by antibiotics.