Clinical Features of Waldenstrom Macroglobulinemia in Korea
Article information
Abstract
Background
Waldenstrom macroglobulinemia (WM) is a lymphoproliferative disorder characterized by monoclonal IgM. Its rarity makes it difficult to know the clinical manifestations and outcomes of patients with WM.
Methods
The clinical records of 13 patients diagnosed with WM between 1983 and 2003 were reviewed, and 12 patients were eligible.
Results
The median age was 57 years (range, 40 to 85), and the male to female ratio was 2. B symptoms and hyperviscosity requiring plasmapheresis existed in 5 and 4 patients, respectively, at the time of diagnosis. Hepatomegaly and splenomegaly were detected in 5 and 3 patients, respectively. Sites of extranodal involvement were bone (3) and lung (1) in 3 patients. The peripheral neuropathy was complicated in 3 patients. (Ed note: check this sentence.) Cryoglobulin was checked in 6 patients and it was detected in 3 of them. The median concentration of serum IgM was 4.2 g/dL (0.7–6.2). The median albumin, hemoglobin, WBC, and platelet levels were 2.8 g/dL, 8 g/dL, 5,400/μL, and 138,000/μL, respectively. One patient had transitional cell carcinoma concomitantly, and one patient developed small cell lung cancer. Of the 11 patients receiving chemotherapy (7-chlorambucil, 2-melphalan, 1-cyclophosphamide, 1-CHOP), 4 patients showed the objective responses including 2 complete remissions, but they all ultimately relapsed. The response rate of second-line therapy was 14% (1/7). After a median follow-up of 20 months, 3 patients were still alive with disease. The median overall and progression-free survival were 24 months (95% confidence interval (CI): 5–43) and 24 months (95% CI: 8–40), respectively.
Conclusion
The initial high levels of serum IgM and severe anemia reflect a lack of suspicion of WM at the early stage. Careful suspicion and proper diagnostic approaches will allow more patients to show an improved outcome.
INTRODUCTION
Since Jan Waldenström first described two patients with Waldenström macroglobulinemia (WM) in 19441), monoclonal IgM gammopathy and lymphoplasmacytic infiltration of the bone marrow have been loosely accepted as diagnostic criteria for this disease. On the WHO classification, WM is considered as a low grade lymphoid neoplasm with a monoclonal IgM, and it is called lymphoplasmacytic lymphoma (LPL)/Waldenstrom macroglobulinemia (WM)2). The immunophenotype of slgM+CD19+CD20+CD5−CD10−CD23−and the cytogenetic abnormality of t(9;14)(p13;q32) provides a histopathologic and molecular basis for the diagnosis of LPL. However, this disease has often been confused with other IgM-secreting lymphoid malignancies. Most of the patients with WM do not have t(9;14)(p13;q32)3), which results in the rearrangement of the PAX-5 gene being reported in up to 50% of all LPL cases4). Thus, Owen RG et al. have proposed alternative diagnostic criteria for WM using the monoclonal IgM gammopathy, bone marrow infiltration and immunophenotyping5).
The age-adjusted incidence rates for WM (per 1 million persons-years at risk) were 3.4 among males and 1.7 among females in the USA6). Although the exact incidence of WM in Korea is not available, its rarity is similar to that of Western countries.
The unsettled diagnostic criteria and the rarity of this disease make it difficult to understand the clinical features and outcomes of patients with WM. Therefore, we conducted this retrospective study to examine the clinical courses of WM in Korean patients.
MATERIALS AND METHODS
The patients reviewed here were selected from all medical records using the disease-specific code system at the Ghil Medical Center and Seoul National University Hospital. The clinical records of 13 patients diagnosed with WM between 1983 and 2003 were reviewed, and 12 patients were found to be eligible for our study. Eligibility was confined to serum monoclonal IgM detected at any level with marrow infiltration of small lymphocytes, plasmacytoid lymphocytes or plasma cells. One patient with insufficient initial data was excluded. Urgent plasmapheresis was performed for the control of symptoms associated with hyperviscosity or neuropathy, and then the physicians recommended that all the patients should undergo systemic chemotherapy.
The association between response rates and each of the variables of interest was measured using Pearson’s chi-square or Fisher’s exact tests. All reported p values are two-sided, and statistical significance was defined as p<0.05. Progression-free survival (PFS) was calculated for all patients from the date of diagnosis until the time of disease progression, relapse, or death, or until the date the patient was last known to be in remission. Overall survival (OS) was defined as the time from induction until death or to the date the patient was last known to be in remission. Response duration was evaluated from the time of complete or partial response until relapse or progression. Actuarial survival and response duration curves were plotted according to Kaplan-Meier’s method, and the prognostic factors were compared using the log-rank test. All calculations were performed upon SPSS for Windows version 10.0.
RESULTS
1) Initial Manifestations
The median patient age was 57 years (range, 40 to 85) and the male to female ratio was 2. Initial clinical and hematologic features are summarized in table 1.
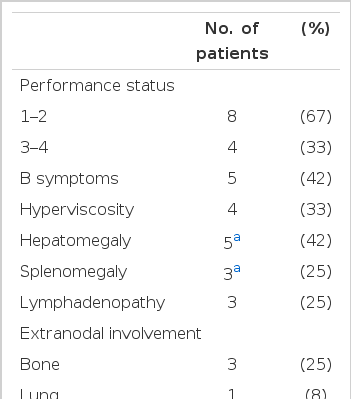
Clinical and hematological features of patients with WM
2) Treatment Outcomes
Plasmaphereses were performed on 3 patients among the 4 patients with the symptoms of hyperviscosity such as dyspnea, or decreased visual acuity due to retinal hemorrhage. Among the three, one patient also had hypercalcemia and another patient suffered from peripheral neuropathy.
One patient refused the chemotherapy and the other 11 patients received the chemotherapy. Seven patients received the chemotherapy with chlorambucil and prednisolone, 2 with mephalan and prednisolone, 1 with cyclophophamide and prednisolone, and 1 with CHOP. Four patients showed responses at a median 3 months-interval after the start of treatment, but ultimately they relapsed. The median duration of the 2 complete and 2 partial responses was 20 months (95% confidence interval (CI): 0–45). The response rates to the second- and third-line therapy were 14% (1/7) and 0% (0/2), respectively. The second line therapy was dexamethasone in 2, fludarabine in 2, melphalan and prednisolone in 1, and BACOP combination therapy in 1 patient, respectively. The third line regimen was melphalan and prednisolone in one and VAD combination in another patient.
After a median follow-up of 20 months, 3 patients were still alive with their disease. Causes of death were as follow: sepsis in 6 patients (pneumonia in 4); second primary malignancies in 2 patients; and uncertain causes in 1 patient. One patient had transitional cell carcinoma concomitantly with the diagnosis of WM, and ultimately the cancer metastasized to the lung. Another patient was diagnosed at an extensive stage of small cell lung cancer after 107 months’ follow-up. In the case whose cause of death was uncertain, cerebrovascular accident was thought to contribute to the patient’s demise. The median OS and PFS were 24 months (95% CI: 4–43) and 24 months (95% CI: 8–40), respectively, for the total patients.
3) Univariate analysis of prognostic factors
Among the sex, age, performance status, anemia, thrombocytopenia, high IgM level and cryoglobulinemia, no variable was associated with an objective response to the first-line chemotherapy. Poor performance status (p=.02), thrombocytopenia (p=.04), and high IgM level (p=.01) were correlated with overall survival. There was no significant prognostic factor for progression-free survival.
DISCUSSION
The consensus panel for clinicopathological definition of WM from the second international workshop on WM in 2002 has recently modified the Owen’s proposal7). According to the new criteria, the diagnosis of WM requires having any level of IgM monoclonal gammopathy and bone marrow infiltration of small lymphocytes showing plasmacytoid/plasma cell differentiation with an intertrabecular pattern. Immunophenotyping is mandatory, but it can be variable within the limits of exclusion of other lymphoproliferative disorders. Our study has several limitations. First, we did not include WM with a nodal presentation. LPL was first defined in REAL (Revised European-American classification of Lymphoid neoplasms). It was previously called lymphoplasmacytic immunocytoma on the Kiel classification and then plasmacytoid small lymphocytic lymphoma on Working Formulation. Most of the cases with nodal presentation of WM could be missed because of the variable registered codes. Another limitation was the impossibility of additional immunphenotyping of marrow specimen the fixation method of marrow biopsy makes it difficult in most cases. Only two of our cases satisfied the above criteria. In a personal communication with Dr. Kim SH from Sungkyunkwan University School of Medicine, he indicated that the frequency of LPL/WM among the lymphoid malignancies was around 0.8%. Eight patients were diagnosed as LPL/WM among the 1,077 patients with B and T lymphoid malignancies in one center during 8 years. To know the exact incidence and clinical features of Korean patients with LPL/WM, further study with the recently defined criteria including immunophenotyping is warranted.
WM patients who do not have symptoms, anemia, significant hepatosplenomegaly or lymphadenopathy, or complications associated with the monoclonal protein may be followed without any treatment until there is evidence of disease progression, just like patients with other low-grade lymphoproliferative disorders. When WM patients present with significant symptoms or signs, prompt treatment of the disease is required. Alkylating agents such as chlorambucil with or without corticosteroids have been regarded as the primary therapy for patients with symptomatic macroglobulinemia8). Response rates to daily and intermittent chlorambucil were about 50%, with both treatments resulting in a median survival of 5.4 years. Among the salvage treatments with nucleoside analogues, both cladribine and fludarabine showed satisfactory responses. Novel therapy with anti-CD20 antibody, or autologous stem cell transplantation could be used as another salvage modality8). Plasmapheresis should be considered as interim therapy for hypervisocity and other IgM-induced complications.
The median survival for patients with WM averages 5 years. At least 20% of patients survive for more than 10 years, and 10–20% die of unrelated causes8, 9). Three multivariate analyses based on a large series have yielded useful prognostic scoring systems10–12). In these systems, only the hemoglobin level had a common prognostic value. Our study failed to show the prognostic significance of anemia on the response to therapy. Other prognostic makers in these systems were age, weight loss, cyroglobulinemia, hypoalbuminemia, number of cytopenia, and serum levels of beta2-microglobulin or IgM.
Schop RFJ3) were the first to document thes pathogenesis of WM at the chromosomal level. The deletion of 6q21 was identified in 42% of WM patients, while translocations involving the immunoglobulin heavy chain (IgH) gene were not found. The translocations involving IgH such as the t (9:14) deregulating PAX-5 gene was associated with IgM-nonsecreting LPL4, 13) and IgM multiple myeloma14). Sahota et al. have guessed at the clonal origin of WM from examining the IgH VDJ sequencing. WM malignant cells transform after cessation of somatic mutation, but without the initiating switch events15).
The median age of WM in Korean patients was relatively younger than that reported in Western studies. High levels of serum IgM and severe anemia reflect the physician’s lack of suspicion of WM at the early stage. The delayed diagnosis and treatment might lessen the response rate and shorten the patient’s survival time. Careful suspicion and detailed diagnostic approaches will allow more of these unique patients to participate in clinical trials and show possible improved outcomes.