Recent insights regarding the molecular basis of myeloproliferative neoplasms
Article information

Abstract
Myeloproliferative neoplasms (MPNs) are a heterogeneous group of clonal disorders characterized by the overproduction of mature blood cells that have an increased risk of thrombosis and progression to acute myeloid leukemia. Next-generation sequencing studies have provided key insights regarding the molecular mechanisms of MPNs. MPN driver mutations in genes associated with the JAK-STAT pathway include JAK2 V617F, JAK2 exon 12 mutations and mutations in MPL, CALR, and CSF3R. Cooperating driver genes are also frequently detected and also mutated in other myeloid neoplasms; these driver genes are involved in epigenetic methylation, messenger RNA splicing, transcription regulation, and signal transduction. In addition, other genetic factors such as germline predisposition, order of mutation acquisition, and variant allele frequency also influence disease initiation and progression. This review summarizes the current understanding of the genetic basis of MPN, and demonstrates how molecular pathophysiology can improve both our understanding of MPN heterogeneity and clinical practice.
INTRODUCTION
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic disorders characterized by the proliferation of differentiated hematopoietic cells [1]. The latest World Health Organization classification of MPN, released in 2017, includes chronic myeloid leukemia (CML), chronic neutrophilic leukemia (CNL), polycythemia vera (PV), primary myelofibrosis (PMF), essential thrombocythemia (ET), chronic eosinophilic leukemia and other, unclassifiable MPNs [2]. MPNs are considered rare diseases, but their incidence rates are increasing [3,4]. According to a recent nationwide population study in Korea, among all MPNs the incidence was highest for ET (2.4 cases per 100,000 population per annum; range, 2.0 to 3.0), followed by PV (1.2 cases per 100,000 population per annum; range, 1.0 to 1.5) and PMF (0.4 cases per 100,000 population per annum; range, 0.3 to 0.5) [5].
MPNs were characterized in 1951 by Dameshek [6] as an overproduction of mature blood cells including erythroid, granulocytic, and megakaryocytic lineages in response to an unknown myelostimulatory factor. He also described the overlap of the clinical and laboratory features of bone marrow hypercellularity, splenomegaly, myelofibrosis progression, and a blast phase [6].
Significant progress in genetic and cellular experimental approaches has allowed for rapid accumulation of molecular data regarding the basis of MPN. It is now known that mutations in tyrosine kinase and other related genes lead to constitutively active proteins, which serve as key myelostimulatory factors [7,8]. These findings have stimulated the development of new targeted treatments, such as Janus kinase (JAK) inhibitors [9]. In the present review, we summarize recent insights regarding the three main mechanisms of MPN pathophysiology: (1) somatic MPN-driver mutations that stimulate activation of JAK-signal transducer and activator of transcription (STAT), (2) cooperating driver mutations in myeloid genes, and (3) other genetic factors involved in the heterogeneity of the MPN clinical phenotype.
KEY SOMATIC DRIVER MUTATIONS IN MPN
The constitutive activation of the JAK-STAT pathway is a hallmark of MPNs [7,8]. Certain somatic driver mutations in hematopoietic stem cells (HSCs) can provide a selective advantage and lead to the formation of clones with JAK-STAT pathway gene mutations and myeloid cell proliferation. Documented drivers include the JAK2 V617F and exon 12 mutations [10-13], mutations in MPL [14], mutations in exon 9 of CALR [15,16], and mutations in the CSF3R (Fig. 1) [17,18]. Recently, the CSF3R mutation was identified as a driver gene in CNL [17]. In addition, negative regulators of the JAK-STAT signaling pathway, including lymphocyte specific adaptor protein (SH2B3, also known as LNK) [19,20], casitas B-lineage lymphoma proto-oncogene (CBL) [21,22], and suppressor of cytokine signaling (SOCS) proteins [23] are inactivated at a low frequency in MPNs, highlighting the importance of the JAK-STAT signaling pathway in MPN pathogenesis.
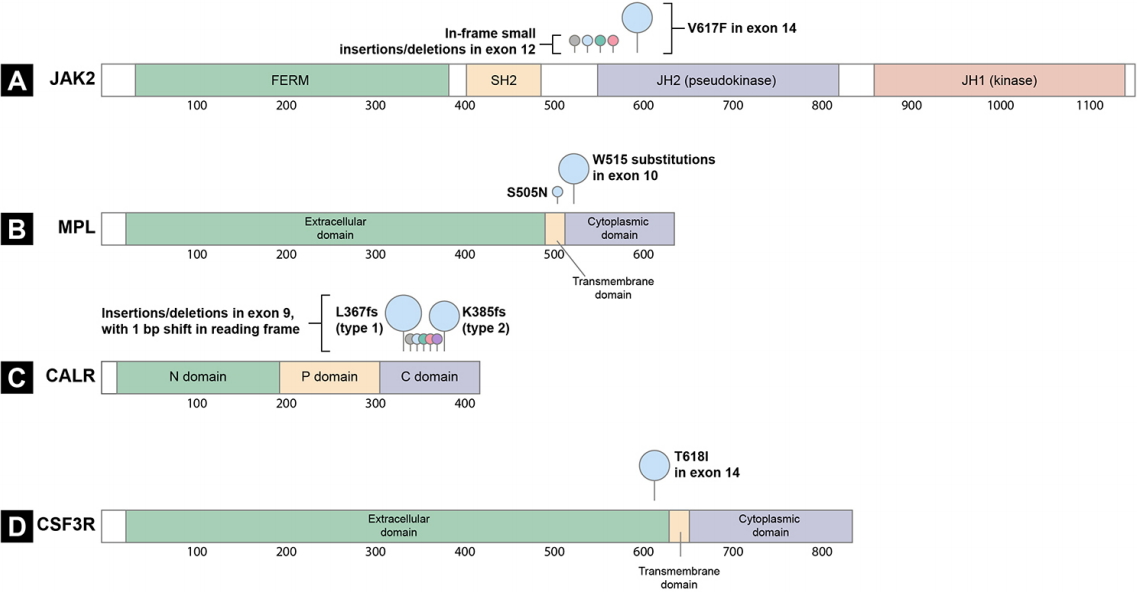
Schematic representation of the protein structure and mutation location. (A) JAK2, (B) MPL, (C) CALR, and (D) CSF3R. The circle size corresponds to the mutation frequency. bp, base pair; C domain, carboxy-terminal domain; FERM, 4.1 protein, ezrin, radixin and moesin; JH, Janus homology; N domain, amino-terminal domain; P domain, proline-rich domain; SH2, Src homology 2.
JAK2 V617F and exon 12 mutations
JAK2, a cytoplasmic tyrosine protein kinase, is required for hematopoietic cytokine receptor signal transduction. The erythropoietin receptor (EPOR), thrombopoietin receptor (TPOR, also known as MPL), and granulocyte colony-stimulating factor receptor (G-CSFR) regulate erythrocytosis, thrombocytosis, and neutrophilia, respectively [24]. Hematopoietic cytokine receptors lack intrinsic kinase activity and thus rely on the associated tyrosine kinase, JAK2 [24]. Normally, interaction of the hematopoietic cytokine receptor with its ligands, such as erythropoietin (EPO), thrombopoietin (TPO), or G-CSF, results in receptor dimerization followed by autophosphorylation and transphosphorylation of the receptor and JAK2 [7]. The activated JAK2-receptor complex then recruits and phosphorylates substrate molecules, including STAT proteins, which leads to target gene transcription in the nucleus (Fig. 2A) [7].

Mutations in JAK2, CALR, MPL, and CSF3R lead to myeloproliferation via constitutive activation of the Janus kinase/ signal transducer and activator of transcription (JAK-STAT) pathway. (A) General mechanism of cytokine activation of the JAK-STAT pathway. Receptor binding leads to dimerization of receptor subunits, inducing transphosphorylation of JAKs. The activated JAKs phosphorylate the receptors, which are subsequently recognized by STATs. STATs are phosphorylated by JAKs and transported into the nucleus, where they regulate transcription of their target genes. Key somatic driver mutations for myeloproliferative neoplasm development occur in (B) JAK2, (C) CALR, (D) MPL, and (E) CSF3R, all of which are associated with JAK-STAT pathway activation.
Two types of JAK2 mutations are associated with MPN (Fig. 1A). The first is a valine to phenylalanine substitution at amino acid position 617 (V617F) in exon 14 of JAK2; this mutation is found in 95% of PV cases and 50% to 60% of ET and PMF cases [10-13]. The second mutation type comprises mutations in exon 12 of JAK2, most of which are in-frame small insertions or deletions [25]. JAK2 exon 12 mutations are observed in 1% to 2% of PV patients, most of whom are JAK2 V617F-negative [25,26]. These mutations are found exclusively in PV and are not associated with ET or PMF [26]. The most frequent mutations are N542-E543del (30%), K539L (14%), E543-D544del (12%), and F537-K539delinsL (10%) [25].
JAK2 mutations promote constitutive activation of the JAK-STAT pathway (Fig. 2B) [10,12]. The V617F mutation, which occurs in the pseudokinase domain (Janus homology 2 [JH2]), leads to loss of the normal auto-inhibitory function of JH2 through changes in JH1 (kinase domain)-JH2 conformation and results in activation of JAK2 [27]. The activated JAK2 mutant recapitulates the physiological response to cytokine binding. Subsequent downstream activation of intracellular signaling occurs via STAT proteins, mitogen-activated protein kinase (MAPK), and phosphoinositidie-3-kinase (PI3K), which play important roles in excessive myeloid cell proliferation and differentiation [27]. Mutations in exon 12 are located upstream of the JH2 domain, within the linker region between the Src homology 2 (SH2) and JH2 domains. JAK2 exon 12 mutations lead to increased phosphorylation of JAK2 and STAT compared to the JAK2 V617F mutations, and result in cytokine-independent activation of JH1 [26]. Patients with the JAK2 exon 12 mutation exhibit more marked erythrocytosis at a younger age [26], suggesting that increased JAK2 promotes the polycythemic phenotype. These differences may reflect differences in the strength of aberrant signaling via the EPOR.
MPL mutations
MPL is a cell surface receptor for TPO [28]. Megakaryopoiesis and platelet production are regulated by TPO, which binds to the cytokine receptor MPL to induce signaling through the JAK-STAT pathway [28]. Gain-offunction mutations in exon 10 of the MPL lead to cytokine-independent growth (Fig. 1B) [28]. MPL mutations at amino acid position 515 (W515) are present in 3% of ET cases and 5% of PMF cases, with the most frequent mutations being W515L and W515K [29]. Several other substitutions at the same position have been reported, namely W515R, W515A, and W515G [30]. A rare germline mutation, MPL S505N, was also described in a hereditary form of thrombocytosis [31].
W515, located in the transmembrane domain of MPL, normally prevents spontaneous activation of MPL [28]. Most substitutions of W515 lead to the activation of MPL in the absence of TPO and result in constitutive activation of JAK2 and STAT protein signaling and cytokine autonomous growth (Fig. 2D) [29].
CALR mutations
In 2013, whole-exome sequencing of MPN samples revealed recurrent somatic mutations in exon 9 of CALR, i.e., the gene that encodes calreticulin [15,16], which is a multifunction calcium-binding chaperone protein [32]. Calreticulin is involved in the regulation of calcium homeostasis and the quality control of newly synthesized glycoproteins in the endoplasmic reticulum (ER) [32].
CALR mutations are the second most common mutation in patients with MPNs (after JAK2 V617F), and are found in 20% to 25% of ET and 25% to 30% of PMF cases [15,33-36]. CALR mutations are rarely reported in PV [15,16]. A knock-in mouse model demonstrated that mutant CALR drives an ET-like and myelofibrosis phenotype, in contrast to JAK2 mutations, which promote a PV-like phenotype [37]. CALR gene mutations shift the reading frame of calreticulin by 1 base pair (bp), resulting in a new C-terminal peptide and the loss of the original KDEL (lysine, aspartic acid, glutamic acid, and leucine) amino acid sequence motif, which plays an important role in retention of calreticulin in the ER [38]. Although calreticulin is not a JAK-STAT signaling molecule, mutant calreticulin interacts with MPL via the new C-terminal peptide to activate downstream signaling (Fig. 2C) [39-43]. Similar to wild-type calreticulin, mutant calreticulin binds to MPL in the ER, but the interaction is reinforced by the positive charges of the new C-terminus [38,43]. Bound by mutant calreticulin, MPL is exported to the cell surface [28]. TPO-independent activation of MPL leads to dimerization and activation of JAK2 and downstream STAT proteins [40]. In addition to binding between mutant calreticulin and MPL, more recent reports showed that three tyrosine residues within the intracellular domain of MPL (Y591, Y626, and Y631), in addition to lectin activity of calreticulin, are also necessary for activation of the JAK-STAT signaling pathway [44].
To date, more than 50 MPN CALR mutations have been found, 80% of which are type 1 or type 2 mutations (Fig. 1C) [34,35]. The type 1 mutation is a 52 bp deletion (c.1092_1143del, p.L367fs*46), whereas the type 2 mutation is a 5 bp insertion (c.1154_1155insTTGTC, p.K385fs*47). The distribution of these CALR mutation types varies depending on the MPN subtype: in PMF, the type 1 mutation is more prevalent than type 2 (70% vs. 13%), but in ET, type 1 and type 2 mutations are similarly distributed (51% vs. 39%) [45]. Recent evidence suggests a relationship between CALR mutation type and MPN clinical features [40,45,46]. Type 1-like mutants are associated with a myelofibrosis phenotype and significantly higher risk of myelofibrotic transformation in ET [46]. In contrast, type 2-like mutants are associated with an ET phenotype, low thrombosis risk (despite higher platelet counts), and indolent clinical course [46]. These mutations showed different structural changes in the exon 9 of calreticulin; the type 1 mutation lost most of the wild-type sequence and calcium-binding sites, whereas type 2 remained closer to the wild-type sequence, retaining about half of the negatively charged original amino acids [15]. These distinctive patterns indicate that the calreticulin mutation type may differentially impair the calcium binding activity of mutant calreticulin [46].
CSF3R mutations
A mutation in CSF3R, which encodes G-CSFR, was recently identified as a CNL driver mutation [17,18]. This mutation activates the receptor, promoting proliferation of mature neutrophils, which are a hallmark of CNL [47]. Differential diagnosis of CNL and atypical CML (aCML), is challenging due to overlapping clinical features such as leukocytosis, splenomegaly, and bleeding diatheses [48]. Thus, the recent discovery of CSF3R mutations has facilitated the differential diagnosis of these disorders. The activating CSF3R T618I mutation is found in more than 80% of CNL patients, but is absent in aCML patients [18]. In 2017, the World Health Organization guidelines were updated to include CSF3R mutations as one of the diagnostic criteria for CNL [49].
The extracellular domain of CSF3R includes a membrane proximal region that is important for granulocytic proliferation, and a cytoplasmic region that regulates granulocytic differentiation and function [18,50]. The most common mutation in CNL, CSF3R T618I, is an activating mutation in the membrane proximal region that results in dimerization of the receptor independent of ligand binding and constitutive activation of the JAK-STAT pathway (Fig. 1D and 2E) [17].
Triple-negative MPNs
Mutations in JAK2, CALR, and MPL are mutually exclusive and account for more than 90% of ET and PMF cases [33]. However, in 10% of ET and 5% to 10% of PMF cases, the disease-driving mutation is unknown, and these cases are referred to as triple-negative [33]. Typical diagnostic workup of MPN patients involves analysis of the JAK2 exon 14, MPL exon 10, and CALR exon 9 for the presence of canonical somatic mutations. However, approximately 10% of triple-negative ET and PMF patients have mutations outside of MPL exon 10 or JAK2 exon 14, and these noncanonical mutations can either be somatically acquired or inherited [51,52]. Noncanonical MPL mutations include T119I, S204F/P, and E230G in the extracellular domain and Y591D/N in the cytoplasmic domain. Noncanonical JAK2 mutations include V625F and F556V [51,52]. Functional studies of MPL and JAK2 noncanonical mutations revealed that they are weak gain-of-function mutations that activate JAK2- STAT signaling [51,52].
COOPERATING DRIVER MUTATIONS IN MYELOID GENES
The known somatic driver mutations in JAK2, MPL, CALR, and CSF3R cannot fully explain the heterogeneity of MPNs. The development of next-generation sequencing has enabled the identification of several mutations that act in concert in more than one-third of MPN patients [53]. These mutations are not specific or restricted to MPN, occurring in other myeloid malignancies including acute myeloid leukemia (AML) [54,55] and myelodysplastic syndrome (MDS) [56,57], as well as in some elderly patients without an overt myeloid malignancy [58-60]. Presence of cooperating mutations in myeloid genes modifies the differentiation process and increases the myelodysplastic features of the disease, implicating continuity among MPN, MDS/MPN, and MDS [7]. The most commonly affected genes are those important in epigenetic regulation, messenger RNA splicing, transcriptional mechanisms, and signal transduction. The gene mutations are not mutually exclusive; two or more mutations can occur simultaneously [53]. The total number of mutations is inversely related to survival and the risk of leukemic transformation in MPN [53]. Cooperating driver gene mutation frequency differs by disease stage; in general, these mutations tend to be more common in patients with progressive disease [61].
Mutations in epigenetic regulator genes
The TET2 and DNMT3A proteins play a central role in the regulation of DNA methylation at cytosine guanine dinucleotide (CpG) sequences [62]. TET2 is involved in active demethylation through oxidation of 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), a process thought to be particularly important for stem cell gene regulation [62]. DNMT3A is responsible for cytosine methylation [63]. All TET2 mutations in MPN are loss-of-function mutations and occur in approximately 10% to 20% cases across MPN subtypes [64]. DNMT3A mutations are less frequent, being present in 5% to 10% of all cases across MPN subtypes [63]. These two types of mutations increase the self-renewal of JAK2 V617F HSCs and play an important role in disease initiation [65]. They also promote disease progression when they occur as secondary mutations, but their role in myelofibrosis and leukemic transformation remains unclear [65].
Mutations in ASXL1 and EZH2, which encode factors involved in histone methylation, are overrepresented in myelofibrosis and AML-transformed disease [53,66]. Mutations in ASXL1 are the second most common epigenetic regulator mutation in MPN after TET2 [8,67]. These mutations occur in approximately 25% of PMF cases [7]. ASXL1 mutations are associated with a poor prognosis and higher frequency of leukemic transformation [68]. A recent study using transgenic mice showed that truncated ASXL1 exerts a gain-of-function effect through interaction with bromodomain and extra-terminal domain (BET) bromodomain-containing protein 4; this finding suggests a new therapeutic approach for targeting ASXL1 mutations in myeloid malignancies [69]. EZH2, a histone methyltransferase of the polycomb repressive complex 2 (PRC2), is involved in the repressive trimethylation of histone H3 at lysine 27 (H3K27) [70]. Loss-of-function mutations in EZH2 have been described in MPNs and result in the de-repression of a set of genes that includes a number of oncogenes (e.g., LMO1 and HOXA9); these mutations are associated with increased HSC self-renewal [70]. EZH2 mutations are relatively common in PMF cases (5% to 10%) and are associated with JAK2 V617F and a poor prognosis [71]. Recent knock-in mice experiments revealed that loss of Ezh2 along with the presence of JAK2 V617F inhibits erythropoiesis and promotes megakaryopoiesis, thereby accelerating the development of myelofibrosis [72].
A number of MPN mutations affect DNA and histone methylation in the HSC compartment, increasing self-renewal and blocking differentiation.
Mutations in mRNA splicing machinery genes
Mutations in spliceosome components, including SF3B1, SRSF2, U2AF1, and ZRSR2, are present in over half of MDS patients [73] and are also seen in MPN patients, especially in ET and PMF cases [66]. The role of these mutations in disease pathogenesis is unclear. Mutations in spliceosome components may alter the splicing of numerous pre-mRNAs and influence functionally relevant mis-spliced events [74,75]. SRSF2 encodes a member of the serine/arginine-rich splicing factor family that binds to exonic splicing enhancer sequences in mRNA [74]. Increasing evidence has shown that SFSR2 mutations affect mRNA motif recognition rather than causing loss of function [74]. Mutations of U2AF1 alter its binding preference to 3′ splice acceptor sites, thereby disrupting the splicing of genes involved in RNA processing and splicing, as well as protein translation [75].
Mutations in transcriptional genes
MPN patients commonly show disrupted transcription factor gene expression. Nuclear factor erythroid 2 (NFE2) is commonly overexpressed in patients with PV, ET, and PMF [76,77]. The molecular pathophysiology of NFE overexpression is associated with JAK2 phosphorylation and JMJD1C demethylation [78]. JMJD1C, a histone demethylase and NFE2 target gene, contributes to NFE overexpression in MPNs through positive feedback. Depletion of JMJD1C selectively reduces JAK2 V617F-mediated cytokine-independent growth, suggesting a potential therapeutic benefit for MPN [78].
Mutations in signal transduction genes
RAS genes (HRAS, NRAS, and KRAS) encode small GTPases with central roles in cell fate signaling pathways [79]. Oncogenic RAS mutations have been identified in a variety of hematologic malignances, such as juvenile myelomonocytic leukemia, chronic myelomonocytic leukemia, AML, and MPN [79]. A recent knock-in mouse study showed that the Kras G12D mutation drives the aggressive MPN phenotype through mediation of Sos1, a guanine nucleotide exchange factor [80,81]. The authors suggested that the survival of patients with KRAS mutations may increase with therapies that target the Sos1-oncogenic Kras interaction [81].
OTHER GENETIC FACTORS AFFECTING THE HETEROGENEOUS CLINICAL MPN PHENOTYPES
Germline predisposition for MPNs
Most cases of MPN are sporadic, but several studies have shown familial clustering of the disease [82-84] and an increased risk of developing MPNs among the relatives of patients [85]. Common predisposing polymorphisms for sporadic MPNs include the germline JAK2 haplotype (GGCC, referred to as “46/1”) and TERT (rs2736100), which confer susceptibility to MPN [86,87]. However, these polymorphisms are relatively common, and few individuals who carry them develop MPN. Recent studies have identified other genetic polymorphisms associated with MPNs operating in diverse cellular processes, including JAK-STAT signaling (SH2B3), DNA cytosine methylation (TET2), transcriptional regulation (GFI1B, PINT), and cell cycle control (CHEK2, ATM) [83,88,89]. These observations have improved our understanding of how mutant clone expansion is influenced by heritable genetic polymorphisms present in the host’s genome [90].
The order of mutation acquisition
The order in which somatic mutations are acquired influences the response to therapy, the biology of stem and progenitor cells, and clonal evolution in patients with MPNs [65,91]. Previous reports demonstrated the importance of mutation acquisition order in JAK2-mutated MPNs harboring concurrent TET2 or DNMT3A mutations [65]. Patients who acquired the JAK2 mutation first have a greater likelihood of presenting with PV than ET, and an increased risk of thrombosis. In contrast, TET2-first or DNMT3A-first patients are more likely to have ET, which accords with the greater self-renewal capacity seen with TET2/DNMT3A mutations compared to JAK2 mutations [65].
Mutant allele burden by MPN subtype
The most frequent mutation, JAK2 V617F, activates the three main hematopoietic cytokine receptors (EPOR, MPL, and G-CSFR) and is associated with PV, ET, and PMF. How the same mutation in JAK2 can occur with three different MPN phenotypes is an important question in MPN research. One possible explanation relates to the mutant allele burden, which is highly variable in granulocytes, with a threshold of detection ranging between 1% and 100% [34]. The mutant allele burden is usually low in ET (around 25%), higher in PV (frequently over 50%), and close to 100% in post-PV or post-ET myelofibrosis [34]. JAK2 V617F has also been detected at very low levels (less than 1%) in the healthy population [88]. JAK2 V617F is one of the most frequent mutations found in age-associated clonal hematopoiesis [58,59].
Leukemic transformation of MPNs
A subset of MPN patients develop secondary AML. Ten years after MPN diagnosis, the rate of leukemic transformation was 1% in ET, 4% in PV, and 20% in PMF [92]. Recent genomic studies have improved our understanding of the somatic mutations that contribute to the transformation of MPN to AML [93-96]. Somatic mutations targeting TP53 tumor-suppressing function are found in 20% to 30% of patients at leukemic transformation but are uncommon in the chronic phase of MPN [93,95]. Transition to leukemia is associated with clonal dominance and transition from mutation heterozygosity to homozygosity [93,95,96]. RUNX1 encodes a transcription factor that binds to enhancers and promoters to regulate normal hematopoiesis [97]. Various RUNX1 mutations and chromosomal aberrations are found in 10% of MPN to secondary leukemia transitions [94,98]. Other signal transduction gene mutations, such as NRAS, SH2B3, CBL, NF1, and FLT3, are associated with an increased risk of leukemic transformation and typically occur more frequently in the blast phase compared to the chronic phase of MPN [7,93,98]. The overall prognosis of patients with leukemic transformation remains quite poor, and treatment is challenging. However, recent advances in gene mutation studies of patients with leukemic transformation are expected to provide mechanistic and therapeutic insights [99].
CONCLUSIONS
The discovery of the molecular features of MPN has revealed novel diagnostic and prognostic markers, provided insight into MPN pathobiology, and prompted new research questions. However, there is much that remain unknown. For example, although several heterogeneous noncanonical mutations in MPL and JAK2 have been identified [51,52], studies have not determined which mutations drive the disease in a significant proportion of triple-negative MPN cases. The hereditary basis of MPNs are poorly understood and future studies should seek to identify novel disease-related germline mutations. In addition, the functional disease outcomes of known germline mutations should be clearly defined. The integration of each of these MPN-related molecular processes into clinical practice is needed to improve and refine disease diagnosis, prognosis, and therapeutics.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was supported by the Soonchunhyang University Research Fund and the Brain Korea (BK) 21 Plus Program.