Rheumatic complications in cancer patients treated with immune checkpoint inhibitors
Article information

Abstract
Immune checkpoint inhibitors (ICIs) have revolutionized anticancer therapy due to their long-term clinical benefits and immune boosting mechanisms. However, despite their consistent therapeutic effects, the use of ICIs is associated with a spectrum of adverse events due to their autoimmune and auto-inflammatory actions. These adverse events can affect any organ system, including the endocrine, neurologic, gastrointestinal, cardiac, skin, pulmonary, and musculoskeletal systems. Of the immune-related adverse events (irAEs), rheumatic complications are common and appear to be distinct from irAEs in other organs in terms of variability of onset time, capacity for persistence, and relationship with pre-existing autoimmune rheumatologic diseases. In this article, we review the mechanisms of the anti-cancer effects of ICIs, the irAEs of immuno-oncology drugs, and the general recommendations for managing irAEs. In particular, we focus on rheumatologic irAEs and discuss their prevalence, clinical characteristics, and management.
CASE PRESENTATION
A 65-year-old man with non-small cell lung cancer (NSCLC) and no history of joint disease presented with a 1-week history of arthralgia in his knees after having received five doses of pembrolizumab (anti-programmed death 1 [PD-1] antibody). He developed pain and swelling in both knees and had difficulty walking. A physical examination revealed tenderness in both knees with a small to moderate degree of effusion. Laboratory studies revealed an elevated erythrocyte sedimentation rate (ESR) of 74 mm/hr (normal range, 0 to 15) and a C-reactive protein (CRP) level of 4.62 mg/dL (normal range, 0.01 to 0.3). Rheumatoid factor (RF), anti-cyclic citrullinated antibodies, and anti-nuclear antibodies (ANA) were negative. How should this patient’s case be managed?
INTRODUCTION
Immune checkpoint inhibitors (ICIs) have recently led to a paradigm shift in various cancer treatments. ICIs against cytotoxic T lymphocyte associated antigen-4 (CTLA-4) and PD-1 have shown clinically significant anti-cancer effects in various cancer types, including melanoma, NSCLC, urothelial cancers, gastrointestinal cancers, and genitourinary cancers [1-4]. The outstanding benefit of ICIs is that they can lead to long-term survival in some subsets of advanced metastatic cancer patients [1,3,5]. This long-term survival benefit is quite rare in the field of cytotoxic chemotherapy and molecular targeted therapy for advanced solid cancers. ICIs revitalize exhausted T-cells by reversing immune tolerance to the cancer cells.
Antigen presenting cells normally elicit an immune response by presenting diverse cancer cell antigens to T-cells. Naïve T-cells are stimulated to convert to cytotoxic T-cells by recognizing tumor antigens with the help of various co-stimulatory ligands and immune cytokines [6]. However, this immune surveillance is jeopardized by immune checkpoints. Immune checkpoints inhibit the overreaction of the immune system that leads to T-cell anergy, exhaustion, and death, so-called immune tolerance [7]. Immune checkpoint (inhibitory) signals play an important role in self-tolerance under normal conditions to prevent hyper-reactive autoimmune responses. Immune tolerance (mediated by immune checkpoints) becomes pathologically predominant in patients with advanced metastatic cancer, resulting in cancer cell proliferation and survival. The major immune checkpoint is CTLA-4 at the antigen presentation stage in dendritic cells (DCs) and PD-1 at the T-cell activation stage. ICIs upregulate immune surveillance against cancer cells by reinvigorating cytotoxic T-cells, resulting in a robust anti-tumor response in advanced solid cancer patients [8]. Despite their considerable anti-cancer effects, ICIs can induce profound inflammatory and immune-related adverse events (irAEs) [9], which can be severe and present challenges for their clinical application. IrAEs can affect almost any organ system, including the endocrine, pulmonary, gastrointestinal, and skin systems (Table 1) [10]. The pattern of these auto-inflammatory and autoimmune toxic effects appears to differ considerably from the side effects of conventional chemotherapeutic agents [2,11], which usually present immune suppressive side effects due to neutropenia.

irAEs from cancer immunotherapy with immune checkpoint inhibitors
Of the irAEs, rheumatic irAEs are commonly underestimated because they present less fatal complications; however, they significantly affect the quality of life of cancer patients and limit the use of ICIs [12]. In this review, the mechanisms of ICIs and irAEs are described, with a special focus on rheumatologic irAEs in terms of their prevalence, clinical characteristics, diagnosis, and treatment.
BALANCE BETWEEN IMMUNE SURVEILLANCE AND IMMUNE TOLERANCE
Immune surveillance, a natural defense mechanism between cancer and the immune system resulting in the elimination of cancer, is a widely accepted phenomenon [13]. Cancer cells initially induce an immune response resulting in the destruction of malignant cells, a process known as immune surveillance. However, immune surveillance fails to recognize the ‘edited’ tumor cells that have escaped surveillance. Immunoediting leads to pro-tumor immunity that blocks anti-tumor adaptive and innate responses and promotes cancer progression. Cancer immunoediting from immune surveillance to immune escape is one of the key phenomena underlying why tumors evade surveillance [14]. Chronic stimulation by malignant cells exhausts T-cells, which are referred to as ‘exhausted T-cells.’ Both innate and adaptive immunity have negative and positive effects on cancer, either by promoting cancer cell survival or by destroying cancer cells. As a result, the immune system has the potential to either promote cancer cell proliferation or cell death. CD8+ T-cells are the principal effector cells of adaptive immunity and immunotherapy, activating and revitalizing these cytotoxic effector T-cells [15]. Increased understanding of immune tolerance by immunoediting will be the foundation for cancer immunotherapy.
MECHANISM OF IMMUNE CHECKPOINT INHIBITORS
ICIs act at two stages of immune checkpoints: T-cell unresponsiveness at the time of activation of naïve T-cells (early stage) and T-cell downregulation and programmed cell death at the effector stage (late stage) [8]. The first immune checkpoint is the CTLA-4 T-cell, which binds the B7 ligand, blocking the costimulatory CD28-B7 interaction during antigen presentation by DCs. CTLA-4 blocks the costimulatory signal leading to T-cell unresponsiveness despite the presentation of tumor antigens [16]. The CTLA-4 inhibitor, an important early ICI, prevents T-cell unresponsiveness and results in T-cell activation [17]. The second immune checkpoint is PD-1, an already activated effector of cytotoxic T-cells [18]. PD-1 binds to programmed death ligand 1 (PD-L1) or programmed death ligand 2 (PD-L2), which are frequently expressed on cancer cells, leading to T-cell exhaustion, anergy, and apoptosis, the main mechanisms of immune escape by cancer cells [19]. Anti-PD-1 or anti-PD-L1 inhibitors act at this second stage, interrupting immune tolerance and leading to T-cell revitalization and stimulation of the ability of T-cells to attack the tumor cells [14,15]. Fig. 1 graphically showed the mechanism of immune check point inhibitors.

Two immune checkpoints of cytotoxic T-cells; targets of immuno-oncology. Two immune checkpoints inhibit T-cell activation. The initial checkpoint is cytotoxic T lymphocyte associated antigen 4 (CTLA-4), which leads to T-cell unresponsiveness despite antigen presentation by dendritic cells (DCs) because it blocks costimulatory interaction between B7-CD28. The subsequent checkpoint, programmed death 1 (PD-1), works after activation of cytotoxic T-cells. PD-1 binds programmed death ligand 1 (PD-L1) and L2, and leads to T-cell exhaustion, anergy, and programmed death of effector cytotoxic T-cells. Cancer cells often overexpress PD-L1 and effectively escape immune surveillance. Anti-CTLA-4 inhibitors and anti-PD-1/PD-L1 inhibitors are the main immune checkpoint inhibitors causing T-cell revitalization and constitute a key mechanism of immuno-oncology. MHC, major histocompatibility complex; TCR, T-cell receptor.
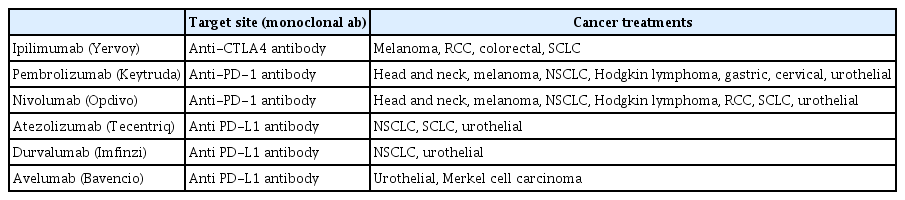
Several ICIs have been approved by the U.S. Food and Drug Administration (FDA) and the Korean FDA: Ipilimumab (Yervoy, Bristol-Myers Squibb, New York, NY, USA) an anti-CTLA-4 antibody, the first agent approved for treatment of advanced melanoma, and pembrolizumab (Keytruda, Merck, Kenilworth, NJ, USA) plus nivolumab (Opdivo, Bristol-Myers Squibb), which are anti-PD-1 antibodies that have been approved for melanoma, metastatic NSCLC, head and neck squamous cell carcinoma, urothelial cancer, gastric adenocarcinoma, mismatch-repair-deficient solid tumors, and classic Hodgkin’s lymphoma. Nivolumab has also been approved for hepatocellular carcinoma and renal cell carcinoma. Pembrolizumab has been approved for PD-L1 positive cholangiocarcinoma, ovarian cancer, and thymic carcinoma. The combination of ipilimumab and nivolumab for advanced melanoma has also received US FDA approval. The PD-L1 antibodies atezolizumab (Tecentriq, Genentech Inc., South San Francisco, CA, USA), durvalumab (Imfinzi, AstraZeneca, Cambridge, UK), and avelumab (Bavencio, Pfizer, New York, NY, USA) have also received approval in the United States. These ICIs, which block CTLA-4, PD-1, and PD-L1, are being rapidly approved for various types of cancer; their indicated use has been extended in Korea and other countries [20]. The field of immuno-oncology is evolving rapidly. Table 2 summarizes the available ICIs and the corresponding types of cancer for which they have been approved according to target site.
Classification of immune checkpoint inhibitors
IrAE MECHANISMS OF IMMUNE CHECKPOINT INHIBITORS
The irAE mechanisms of ICIs can be grouped into three categories: (1) The tumor antigen itself is a self-antigen, so the ICIs attack not only cancer cells but also normal cells. (2) Epitope spreading (determinant spreading); cancer cells secrete both tumor-associated antigens (neo-antigens) and self-antigens. The initial immune response attacks tumor-associated antigens (initial dominant epitope), after which it broadens and diversifies immunity to self-antigens (secondary endogenous epitope), resulting in an auto-inflammatory response to self-antigen-containing normal cells, which is another important mechanism of chronic autoimmune or inflammatory responses [21]. (3) The anti-CTLA-4 antibody downregulates regulatory T-cells (Treg), leading to immune revitalization and attack of normal self-cells [22-24]. However, the exact irAE mechanisms have not been fully elucidated. Although ICIs are usually less toxic than traditional chemotherapy, the toxicology profile of immuno-oncological drugs differs considerably from traditional chemotherapy immunosuppressive complications, especially neutropenic infections, and these drugs display a new spectrum of adverse events of autoimmune or auto-inflammatory origin—the so-called irAEs [9,13]. It is imperative that clinicians are aware of the unique toxicity profiles of ICIs with rapidly increasing use. IrAEs have been reported in almost every organ system, including the gastrointestinal, endocrine, respiratory, skin, rheumatic, renal, and neural systems. Table 1 summarizes the irAEs reported in each organ. IrAEs can occur in a single organ or in several organs by the process of immune activation [11].
GENERAL ASPECTS OF irAEs
The onset time of irAEs is within 3 months from the start of ICI use. However, irAEs can occur even 1 year after the discontinuation of immuno-oncology therapy [10]. Although there is no specific role for the pathologic diagnosis by biopsy when diagnosing irAEs, accurate diagnoses are necessary to select appropriate treatment strategies for Grades 3 and 4 severe cutaneous, gastrointestinal, renal, and pulmonary irAEs. Careful patient selection, baseline assessment of underlying autoimmune disease, and collection of family history are necessary before initiating treatment with ICIs. Awareness and early diagnosis are essential for the management of irAEs, including cessation of ICIs and timely use of glucocorticoid treatment [25]. High-dose steroids, tumor necrosis factor-α (TNF-α) blockers, mycophenolate, and tacrolimus should be considered to manage irAEs depending on the grade and organ involvement. A meta-analysis revealed that the prevalence of irAEs is up to 75% for anti-CTLA-4 [26] and close to 30% for anti-PD-1 and/or anti-PD-L1 therapies [27]. Some patterns of toxicity are associated with specific immunotherapy drugs or cancer types. Anti-CTLA-4 antibodies are associated with more severe toxicities than anti-PD-1/PD-L1 antibodies: up to 43% of patients treated with ipilimumab (anti-CTLA-4 antibody) suffer from Grade 3 or more toxic events, compared with less than 20% of patients treated with anti-PD-1 or anti-PD-L1 antibodies. Although most irAEs are mild to moderate, fatal complications can occur in up to 2% of patients, mostly caused by severe pneumonitis, fulminant bowel disease, and more rarely by myocarditis, encephalitis, toxic epidermal necrolysis, and type 1 diabetes with acute ketoacidosis. Of the irAEs, rheumatic complications are common, but are difficult to diagnose and have quite distinct characteristics from irAEs of other organ systems, in terms of variable onset time and capacity to persist even after cessation of ICIs [12,28]. This review considers the prevalence, clinical characteristics, and treatment approaches for rheumatic irAEs.
MUSCULOSKELETAL IMMUNE-RELATED ADVERSE EVENTS
Musculoskeletal symptoms, such as arthralgia and myalgia, are likely to occur in 5% to 16% of cancer patients undergoing treatment with ICIs [29], whereas severe or life-threatening musculoskeletal irAEs are rare. Clinician awareness of musculoskeletal irAEs is relatively poor, but musculoskeletal irAEs can have a significant effect on functional activities, leading to a reduced quality of life. This review focuses on the epidemiology, clinical manifestations, and management of the most frequent musculoskeletal irAEs, which are inflammatory arthritis, myositis, and polymyalgia rheumatica (PMR)-like syndrome [28].
Epidemiology
The incidence of true musculoskeletal irAEs is not clearly defined. There are two main causes for the discrepancies in reports of the incidence of musculoskeletal irAEs. First, the codes for the musculoskeletal adverse events used in oncology clinical trials are inconsistent. For example, arthritis has been coded as arthralgia, joint effusion, or musculoskeletal pain. This discordance could make understanding of the incidence of musculoskeletal irAEs difficult. The second reason is the grading system used in oncologic clinical trials concerning adverse events. Most clinical trials do not report more than Grade 3 adverse events, according to the oncologic Common Terminology Criteria for Adverse Events (CTCAE), in which irAEs are graded from 1 to 5 (1 = mild, 2 = moderate, 3 = severe, 4 = life threatening, and 5 = death related to toxicity). However, severe adverse events (Grades 3 to 5) leading to disability or requiring hospitalization are rare with musculoskeletal irAEs. The application of the same CTCAE grading system to musculoskeletal irAEs thus appears to be inappropriate. Therefore, the true incidence of musculoskeletal irAEs is limited [12,28].
No prospective observational study has systematically assessed musculoskeletal irAEs. Although some retrospective observational studies have reported the incidence of musculoskeletal irAEs, detailed clinical, laboratory, and imaging information is limited. The reported incidence of inflammatory arthritis as an irAE ranges from 0.7% to 5.1% [30-32] The reported incidence of myositis induced by ICIs (irMyositis) ranges from 0.15% to 1.28%, and the incidence could be higher for the combination of anti PD-1 and anti-CTLA4 inhibitors than for monotherapy [33,34]. Although the incidence of PMR induced by ICIs (irPMR) has not been well studied, one study reported that the frequency of irPMR was 2.09% [35].
Clinical characteristics
Inflammatory arthritis
Inflammatory arthritis comprises a group of diseases characterized by inflammation of the joints [34]. The presence of inflammation at joints can be identified by any of the four cardinal signs of inflammation (erythema, warmth, pain, and swelling), morning stiffness (lasting more than 30 min to 1 hour) or increased levels of inflammatory markers, such as ESR or CRP. The clinical manifestations of inflammatory arthritis resulting from ICIs are variable. To date, two subtypes of inflammatory arthritis have been mainly described: a subtype of polyarthritis similar to rheumatoid arthritis usually affecting the small joints (proximal interphalangeal, metacarpal, and wrist); and a subtype resembling spondyloarthritis (SpA) characterized by inflammatory back pain, enthesitis, dactylitis, and predominantly large joint involvement [28,29,34,36]. In the spectrum of SpA, reactive arthritis (conjunctivitis, sterile urethritis, and oligoarthritis) and psoriatic arthritis have also been reported [35,37,38]. The median onset time for the development of inflammatory arthritis ranges from 2 to 24 months after ICI exposure [32,39-41]. One retrospective analysis found that the median time until the resolution of arthralgia after the last dose of immunotherapy was 9.2 ± 6.1 months, and musculoskeletal symptoms could last more than 1 year [42].
Autoimmune serology of most patients with inflammatory arthritis is negative for RF, anti-citrullinated protein antibodies (ACPA), and ANA. Imaging studies such as ultrasound and magnetic resonance imaging (MRI) have confirmed inflammatory arthritis, which involves synovitis with or without an increased Doppler signal, joint effusion, tenosynovitis, and bony erosion. Joint fluid analysis has been performed in several cases, revealing inflammatory characteristics (2,000 to 50,000 white blood cells/μL with neutrophil predominance) [28,39,43].
Myositis
Inflammatory myositis is a heterogeneous group of chronic conditions characterized by muscle inflammation associated with motor weakness [44]. Clinical manifestations of irMyositis have been consistent with necrotizing myopathy or polymyositis, whereas dermatomyositis (DM) is rarely reported. The clinical phenotypes and severity of irMyositis range from isolated oculomotor dysfunction to fulminant myositis of the heart and diaphragm [33,45-47]. Clinical features are myalgia, proximal muscle weakness, ptosis, and oculomotor weakness with diplopia. Compared to idiopathic inflammatory myositis, irMyositis has a more sudden onset of symptoms (usually in the first 2 months of ICI treatment). The onset of symptoms varies from 5 days to 115 weeks [33,40,45,46,48]. Extra-ocular muscles are spared even in advanced or untreated cases of inflammatory myositis. However, irMyositis is often accompanied by oculomotor weakness with diplopia resembling ocular myasthenia gravis (MG); a subset of patients with irMyositis also has concomitant MG [45,46,49]. One study found that up to 32% of irMyositis cases were complicated by concomitant myocarditis [46]. The reported incidence of myocarditis is 0.15% for treatment with nivolumab and 0.24% for the combination treatment with nivolumab and ipilimumab. In cases of fulminant myocarditis, the development of myositis, very high levels of troponin I, early progressive and refractory cardiac electrical instability, and mortality were noted despite the administration of high-dose glucocorticoids and supportive care [33].
Serum levels of creatinine kinase (CK) tend to be markedly increased, by a factor of 10 or more. In most cases, the myositis-specific antibodies (MSAs) and myositis-associated autoantibodies (MAAs) are negative. In most cases, electromyography (EMG) reveals a myopathic pattern characterized by short duration, low amplitude polyphasic units on voluntary activation, presence of abnormal spontaneous activity, and complex repetitive discharges. Muscle biopsies show a variable degree of necrotizing myopathic changes with infiltrates of macrophages in the endomysium and perimysium. Findings of muscle major histocompatibility complex I (MHC-I) expression and infiltration of CD8+/CD4+ T-cells have been reported to varying degrees [45,46,50].
PMR-like syndrome
PMR is an inflammatory disorder characterized by severe pain and stiffness affecting the shoulders and pelvic girdle bilaterally. Giant cell arteritis (GCA) is a large vessel vasculitis characteristically affecting one or more branches of the carotid artery, particularly the temporal artery. PMR and GCA represent different clinical manifestations of the same disease process [51]. PMR-like syndrome is one of the major clinical phenotypes of musculoskeletal irAEs accompanied with inflammatory arthritis. PMR-like syndrome alone is more common than the concurrent occurrence of PMR and GCA. The time to onset of PMR/GCA after exposure to ICIs ranges from 10 days to 1 year [35,52,53]. The clinical manifestations of irPMR are similar to those of the traditional forms of PMR, such as pain and stiffness in bilateral shoulders and the pelvic girdle. In cases of GCA induced by ICIs (irGCA), symptoms include headache, scalp tenderness, jaw claudication, and transient diplopia with one episode of amaurosis fugax, consistent with the typical form of GCA [52]. Clinical improvement in response to corticosteroids (7 to 60 mg/day) is achieved rapidly in most cases [35,52,53].
Serum CRP levels tend to increase in patients with irPMR/irGCA, as increases in acute phase reactants are classic clinical features of PMR/GCA. However, one study found that 36.4% (four of 11 patients) of patients with typical clinical features of PMR did not have any increase in CRP [35]. In most cases, RF and ACPA were negative [35,52-54]. Ultrasound revealed subdeltoid bursitis, biceps tenosynovitis, glenohumeral synovitis, hip synovitis, or trochanteric bursitis, which were included in the 2012 European League against Rheumatism and American College of Rheumatology provisional classification criteria [55]. Histopathological findings of the temporal artery in patients with irGCA also correspond with the findings of the classic form of GCA, which involves intima proliferation and fragmentation of the internal elastic lamina [52].
Management of irAEs
General recommendation for irAEs according to grade
Although the National Comprehensive Cancer Network and American Society of Clinical Oncology (ASCO) guidelines present recommendations for specific organ-based toxicity diagnosis and management that vary according to the organ system affected, they also present general guidelines for the management of irAEs according to severity [10].
(1) Grade 1 (mild toxicities): Exclude specific neurologic, hematologic, and cardiac toxicity, and continue treatment under close monitoring.
(2) Grade 2 (moderate toxicities): Temporarily discontinue the use of ICIs until laboratory tests or clinical symptoms show that the reaction is improved; corticosteroids can be a replacement therapy with a starting dose of prednisone at 0.5 to 1 mg/kg/day.
(3) Grade 3 (severe toxicities): Administer high-dose corticosteroids (prednisone at 1 to 2 mg/kg/day or methylprednisolone at 1 to 2 mg/kg/day) for at least 4 to 6 weeks; extreme caution is advised in cases of restarting immunotherapy, if the physician in charge decides to continue with this treatment. Some refractory cases may require infliximab or other immunosuppressive agents.
(4) Grade 4 (very severe toxicities): Immediately and permanently cease administration of checkpoint inhibitors with the exception of endocrine diseases previously controlled by hormone replacement therapy.
Recommendations for musculoskeletal irAEs
A multidisciplinary approach is needed to manage patients with irAEs. Two main concerns for clinicians relate to how to manage various phenotypes of irAEs and whether to maintain ICIs in cancer patients with irAEs. A management algorithm for irAEs has been outlined by a multidisciplinary working group, and subsequent guidelines from the European Society for Medical Oncology (ESMO) and the ASCO have been published for the diagnosis and treatment of irAEs [10,34,56,57]. Due to limitations in the available evidence, the guidelines are based on informal consensus [10,57]. These treatment guidelines are based on the severity and involved organ systems of the irAEs. Although specific recommendations for treating inflammatory arthritis have been presented in both the ESMO and ASCO guidelines, those for myositis and PMR-like syndromes have been included only in the ASCO guidelines. The potential effects of steroids, synthetic, or biologic disease-modifying anti-rheumatic drugs (DMARDs) for managing irAEs on tumor progression have not been fully determined. However, recent studies suggest these treatments are not associated with risk of cancer progression, recurrence, or survival [43].
1) Inflammatory arthritis
Recommended diagnostic algorithms for inflammatory arthritis by ESMO and ASCO are listed below [10,57]. Early recognition of the disease and involvement of rheumatologists is important, as erosive joint damage is seen within weeks of symptom onset.
(1) Grade 1 (mild pain with inflammation, erythema, or joint swelling): History and examination of joints (swelling/tenderness/range of motion), spine, and skin; consider plain X-ray/ultrasound/MRI to exclude metastases and evaluate joint damage (erosions); consider autoimmune blood panel (ANA, RF, anti-cyclic citrullinated peptide [anti-CCP]), and inflammatory markers (ESR, CRP); consider testing human leukocyte antigen-B27 if symptoms are suggestive of SpA.
(2) Grade 2 (moderate pain associated with the symptoms of Grade I, limited instrumental activities of daily living [ADL]): Complete history and physical examination, laboratory tests as in Grade I; consider musculoskeletal ultrasound and/or joint MRI when clinically indicated (e.g., unresponsiveness to treatment, need for differential diagnosis between metastases or septic arthritis); consider early rheumatology referral if symptoms persist.
(3) Grades 3 to 4 (severe pain associated with the symptoms above; irreversible joint damage disabling, limited self-care [ADL]): As with Grade 2, seek rheumatologist advice and review.
Recommended treatment algorithms for inflammatory arthritis by ESMO and ASCO are as follows. Compared to the general recommendations for irAEs, a lower dose of systemic steroids is required for inflammatory arthritis. Additionally, intra-articular steroid injections are one of the treatment options for large joints.
(1) Grade 1: Initiate analgesia with acetaminophen and/or nonsteroid anti-inflammatory drugs (NSAIDs).
(2) Grade 2: Withhold ICIs temporally and resume when symptoms improve with prednisolone ≤ 10 mg/day; escalate analgesia and consider higher doses of NSAIDs as needed; consider intra-articular steroid injections for large joints; if the disease is inadequately controlled, initiate prednisolone 10 to 20 mg/day or equivalent for 4 to 6 weeks; if there is improvement, slowly taper during the next 4 to 6 weeks; if no improvement is seen after the initial 4 to 6 weeks, treat as in Grade 3; if it is not possible to lower corticosteroid dose to < 10 mg/day after 3 months, consider DMARDs.
(3) Grades 3 to 4: Withhold ICIs and may resume in consultation with rheumatology, if recover to Grade 1 or less; initiate oral prednisone at 0.5 to 1 mg/kg; in case of failure of improvement after 4 weeks or worsening in the meantime, consider synthetic DMARDs (methotrexate, leflunomide, sulfasalazine) or biologic DMARDs (TNF-α or interleukin 6 [IL-6] inhibitor).
2) Myositis
Recommended diagnostic evaluations by ASCO for irMyositis are as follows [10]. Awareness of accompanying myocarditis is important, otherwise it could lead to mortality.
(1) Grade 1 (mild weakness with or without pain): History, neurologic examination, including muscle strength, examination of the skin for findings suggestive of DM; laboratory testing including creatine phosphokinase (CK), lactate dehydrogenase, transaminases, aldolase, ESR, CRP; troponin, and other cardiac tests, including echocardiogram, as needed to evaluate myocardial involvement; consider EMG, MRI, and/or muscle biopsy as appropriate when diagnosis is uncertain; consider paraneoplastic autoantibody testing for myositis and neurologic conditions, such as MG.
(2) Grade 2 (moderate weakness with or without pain, limited age-appropriate instrumental ADL): Complete history, physical examination, and laboratory tests as in Grade 1; autoimmune myositis panel (MSA and MAA), EMG, and MRI for affected joints; consider muscle biopsy if diagnosis is uncertain, early referral to a rheumatologist or neurologist.
(3) Grades 3 to 4 (severe weakness with or without pain, limited self-care ADL): As for Grade 2, urgent referral to a rheumatologist or neurologist.
Recommended treatment algorithms by ASCO for irMyositis are as follows [10].
(1) Grade 1: Offer analgesia with acetaminophen and/or NSAIDs; if CK is elevated and patient has muscle weakness, treat as Grade 2.
(2) Grade 2: Withhold ICIs temporally and resume when symptoms improve, if CK is normal and on prednisolone < 10 mg/day; NSAIDs as needed; if CK is elevated (three times or more), initiate prednisolone at 0.5 to 1 mg/kg/day; permanent discontinuation of ICIs may be required in the majority of patients with Grade 2 symptoms and objective findings (elevated muscle enzymes, abnormal EMG, MRI, or biopsy).
(3) Grades 3 to 4: Withhold ICIs until Grade 1 or less while off immune suppression and permanently discontinue if any evidence of myocardial involvement; initiate prednisone 1 mg/kg; consider 1 to 2 mg/kg/day of intravenous methylprednisolone or higher dose bolus if severe compromise (severe weakness limiting mobility, cardiac and respiratory insufficiency, dysphagia); consider plasmapheresis; consider intravenous injections of immunoglobulins; consider other immunosuppressant therapy (methotrexate, azathioprine, or mycophenolate mofetil) if failure of improvement after 4 to 6 weeks or worsening.
3) PMR-like syndrome
Recommended diagnostic evaluations by ASCO for irPMR are as follows [10].
(1) Grade 1 (mild stiffness and pain): Complete rheumatologic history regarding differential diagnosis and examination of all joints and skin; check for symptoms of GCA, such as headache or visual disturbances; refer to ophthalmologist if symptoms are present, and consider temporal artery biopsy; laboratory testing including ANA, RF, anti-CCP, ESR, CRP, and CK.
(2) Grade 2 (moderate stiffness and pain, limiting age-appropriate instrumental ADL): As for Grade 1; early referral to a rheumatologist.
(3) Grades 3 to 4 (severe stiffness and pain, limiting selfcare ADL): As for Grade 2; seek rheumatologist advice and review.
Recommended treatment algorithms for irPMR by ASCO are noted as follows [10]. Treatment for ≥ Grade 2 irPMR is according to the general recommendations for classic PMR [51].
(1) Grade 1: Offer analgesia with acetaminophen and/or NSAIDs.
(2) Grade 2: Withhold ICIs temporally, and resume upon symptoms improved, prednisolone < 10 mg/day; initiate prednisolone at 20 mg/day or equivalent; if improvement is noticed, start to taper after 3 to 4 weeks; if there is no improvement after the initial 4 weeks, treat as Grade 3.
(3) Grades 3 to 4: Withhold ICIs and may resume in consultation with rheumatology, if patient recovers to Grade 1 or less; initiate prednisone 20 mg/day; if failure to improve, consider a steroid sparing agent such as methotrexate or IL-6 inhibitor.
TUMOR RESPONSE ACCORDING TO IRAES
The development of irAEs during ICI treatment suggests shifting from the exhausted T-cell phenotype to an active effector T-cell phenotype. However, the question of the relationship between the occurrence of irAEs and the anti-tumor response to ICI treatment remains unanswered. A prospective cohort study from a single center found that patients experiencing rheumatic or other type of irAEs had a higher response rate to ICI treatment compared to those without [35]. However, two retrospective studies yielded conflicting results. One study found an association between cutaneous irAEs and overall survival in melanoma patients treated with nivolumab [58], while the other found that overall survival was not affected by occurrence of irAEs in melanoma patients treated with ipilimumab [12]. Further studies are needed to clarify the association between irAEs and the tumor response to ICIs.
ICIS IN PATIENTS WITH PRE-EXISTING AUTOIMMUNE DISEASES
Data on the safety of ICIs in patients with cancer and underlying autoimmune diseases are limited, and guidelines or recommendations for these patients are missing. Patients with preexisting autoimmune diseases were excluded from the initial clinical trials for FDA approval of ICIs [12]. Several retrospective studies found that 6.25% to 55% patients with autoimmune diseases who were treated with ICIs, experienced disease flares, and another 16% to 22% developed new irAEs while receiving ICI therapy. Most of these irAEs were mild to moderate (CTCAE Grades 1 to 2) and were treated with glucocorticoids. The reported incidence of irAEs ≥ 3 ranges from 10% to 33%, yet most of these severe irAEs were also treated by discontinuing ICIs and glucocorticoid ± immunosuppressant agents [59-63]. Overall survival or tumor response to ICIs did not differ significantly in patients with pre-existing autoimmune diseases [62,63]. Recent retrospective studies have provided evidence for the feasibility of ICIs for patients with pre-existing autoimmune diseases. Largescale prospective studies are required to validate these findings and establish evidence based on guidelines for ICI therapy in patients with coexisting cancer and autoimmune diseases.
MANAGEMENT AND CLINICAL PROGRESS OF THE PRESENTED CASE
In the vignette, we described a man with Grade 2 inflammatory oligo-arthritis involving large joints as irAEs, who presented at our institution. Imaging studies were performed. Plain X-rays of the knee joints revealed no radiographic joint damage. Ultrasound revealed joint effusion and synovial hypertrophy with an increased power Doppler signal. Subsequently, arthrocentesis was performed for the differential diagnosis, such as crystal-induced arthritis and septic arthritis. The synovial fluid was inflammatory (white blood cell count was 25,900/mL, neutrophils 76%, lymphocytes 5%) with no evidence of crystals, and the culture results were negative. Without initiating prednisolone or discontinuing the ICI, inflammatory arthritis improved with intra-articular corticosteroid injections and NSAIDs for 2 weeks. After 10 doses of pembrolizumab, inflammatory arthritis recurred in his elbows, which subsequently responded to 10 mg/day prednisolone with intra-articular corticosteroid injections. The arthritis improved significantly 2 weeks later. The patient remains in durable stable disease without a flare of inflammatory arthritis 12 months after initiating pembrolizumab.
CONCLUSIONS
The development of immuno-oncology drugs such as anti-PD-1 and anti-CTLA-4 inhibitors has led to a paradigm shift in the treatment of cancer patients. The revolutionary anti-cancer effects and the wide spectrum of indications of ICIs, regardless of cancer type, have increased their use exponentially. As their utilization expands, there is a great likelihood of an increase in the frequency of irAEs, which have been relatively underestimated. The mechanism, clinical characteristics, and pattern of management are quite different from managing the adverse events of cytotoxic chemotherapy. The occurrence of various irAEs can limit the use of ICIs and lead to fatal cancer progression. Timely screening and early detection of irAEs are critical to manage irAEs and long-term use of immune oncology drugs. Rheumatic irAEs are some of the most frequently encountered irAEs and could be severe and life-threatening. Awareness of rheumatic irAEs is an important first step, and a multidisciplinary approach is vital for their management.
Notes
No potential conflict of interest relevant to this article was reported.