Diagnosis and treatment of cystic lung disease
Article information
Abstract
Cystic lung disease (CLD) is a group of lung disorders characterized by the presence of multiple cysts, defined as air-filled lucencies or low-attenuating areas, bordered by a thin wall (usually < 2 mm). The recognition of CLDs has increased with the widespread use of computed tomography. This article addresses the mechanisms of cyst formation and the diagnostic approaches to CLDs. A number of assessment methods that can be used to confirm CLDs are discussed, including high-resolution computed tomography, pathologic approaches, and genetic/ serologic markers, together with treatment modalities, including new therapeutic drugs currently being evaluated. The CLDs covered by this review are lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, Birt-Hogg-Dube syndrome, lymphocytic interstitial pneumonia/follicular bronchiolitis, and amyloidosis.
INTRODUCTION
A pulmonary cyst is an air-filled lucency or low-attenuating area bordered by a thin wall (usually < 2 mm) and having a well-defined interface with normal lung tissue [1]. In Korea, clinical awareness of cystic lung disease (CLD) has recently increased due to the widespread use of high-resolution computed tomography (HRCT) and the country’s general-health medical examinations [2]. Cysts are rare in asymptomatic individuals < 55 years of age but their prevalence increases with age [3]. CLD is a constellation of diverse lung disorders that originate from various causes but they have in common an anatomical cystic deformity. Due to its etiological heterogeneity, CLD can be classified based on the underlying pathophysiologic mechanisms: congenital, genetic, infectious, inflammatory, lymphoproliferative, neoplastic, and smoking-related [4,5]. CLD must also be distinguished from emphysema, cavity, bulla, bleb, pneumatocele, and honeycombing, all of which feature lucencies that mimic a pulmonary cyst. These can be differentiated from pulmonary cysts based on the thickness of the wall, the size of the gas-filled space, the anatomic location, and the tendency of true cysts to cluster [1]. Table 1 summarizes the cyst and cyst-mimicking changes seen on chest radiography and/or HRCT.

In this review, we discuss the mechanisms of cyst formation and the evaluation of CLDs. We will also present a number of methods used to confirm CLD. Our review includes the following disorders: lymphangioleiomyomatosis (LAM), pulmonary Langerhans cell histiocytosis (PLCH), Birt-Hogg-Dube (BHD) syndrome, lymphocytic interstitial pneumonia (LIP)/follicular bronchiolitis (FB), and amyloidosis.
CYST FORMATION
The pathogenesis of cyst formation in the lung is unclear. Several mechanisms have been suggested based on various pathophysiologic processes, and specific diseases have been linked to each of the proposed mechanisms. Check-valve obstruction with subsequent distal over-inflation is one of the most widely known pathways leading to CLD and has been linked to diseases such as FB, metastatic neoplasm, pneumatocele, LAM, and PLCH [6-10]. However, the cysts in LAM or PLCH become smaller in expiratory computed tomography (CT) scans, which suggests that these cystic lesions communicate to some degree [11]. Ischemia is another process that can induce cystic change in the lungs. The obstruction of small capillaries that supply the terminal bronchiole leads to necrosis of the airways and ischemic dilatation [12]. A molecular mechanism has also been suggested in which matrix metalloproteinase (MMP), matrix-degradation enzymes, and podoplanin (D2-40) induce lung tissue remodeling [9,13,14]. LAM, PLCH, and light chain deposit disease (LCDD) are representative diseases that develop via this mechanism.
Finally, genetic mutation is thought play a role in cyst formation, especially in BHD syndrome. The pathologic findings in pure BHD syndrome (without pneumothorax) include cysts that mostly abut interlobular septa, without significant inflammation, suggesting that the abnormality is at the alveolar-septal junction. BHD syndrome has also been linked to a folliculin (FLCN) gene mutation [15]. Immunohistochemical study of FLCN expression in lung tissue from these patients shows poorly colocalized surfactant protein C (SP-C) expression in alveolar epithelial cells. The loss of FLCN function in a mouse model of BHD syndrome leads to increased alveolar epithelial cell apoptosis and alveolar enlargement, suggesting a role for FLCN in cyst formation [16].
DIAGNOSTIC APPROACHES
Initial discovery of a cystic change on a patient’s lung is followed by noting its characteristic radiological findings. HRCT depicts several relevant aspects of cystic lesions, such as their distribution (diffuse/random, or confined to a specific region), size, and shape (round or bizarre, or lentiform). While the cysts in LAM, LIP, FB, and amyloidosis show a diffuse or random distribution, those in PLCH typically have an upper/middle zone predominance and the cysts in BHD syndrome have a basilar/peripheral/subpleural zone distribution. The thickness of the cyst wall is another diagnostically useful feature. Protruding or solid internal structures in the pulmonary cyst are suggestive of FB, LIP, or BHD. Aside from the cyst itself, other informative radiographic findings are pleural effusion, parenchymal nodule, and ground glass opacity (GGO) [8,17-19].
The recognition of an abnormal lung parenchymal cystic foci is frequently followed by histologic confirmation via adequate tissue procurement, such as a bronchoscopic biopsy, transbronchial lung biopsy, video-assisted thoracoscopic surgery biopsy, or even open lung biopsy, depending on the patient’s condition and the diagnostic advantages. On conventional microscopic hematoxylin-eosin staining, true cysts have an epithelial cell lining, whereas a pseudocyst has no true cell covering [19]. As previously described, cystic lesions may exhibit a tropism in terms of their distribution. The cysts in BHD syndrome are distributed in the intraparenchymal or basilar region, whereas those in emphysemas are located at the apex or have a centrilobular distribution. On microscopic examination, LAM, PLCH, and neoplasms contain abnormal cellular proliferations that require special staining and/or immune histochemical assessment. Chronic hypersensitivity pneumonitis and connective tissue disease-interstitial lung disease characteristically show chronic inflammatory infiltration and fibrosis. In LAM, the detection of human melanoma-black 45 (HMB-45), and in PLCH patients the expression of CD1A, aid in the diagnosis. Apple-green birefringence in tissues stained with Congo red is important for differentiating pulmonary amyloidosis from LCDD [9,19]. In addition to radiologic and pathologic evaluation, positivity for biomarkers such as vascular endothelial growth factor D (VEGF-D) and MMPs, the verification of genetic alterations, and the presence of concomitant disease are useful diagnostic features [20- 22].
CAUSES OF CYSTIC LUNG DISEASE
Lymphangioleiomyomatosis
This multisystemic, progressive disorder primarily affects the lungs of females of childbearing age (median age of 35) [23]. LAM can develop both sporadically (S-LAM) and genetically in patients with tuberous sclerosis complex (TSC-LAM). TSC, an autosomal dominant trait, is a neurocutaneous disorder characterized by multiple benign hamartomatous lesions in the brain, skin, kidney, heart, and eye [24]. LAM occurs in 30% to 40% of females and 10% to 15% of males with TSC. However, the cystic changes specific to LAM with TSC are usually asymptomatic in males [4,24]. S-LAM occurs almost always in females.
Both S-LAM and genetic LAM are associated with mutations in tumor suppressor genes, including TSC1 (encoding hamartin) and TSC2 (encoding tuberin) [20]. Mutations in TSC1 or 2 result in the abnormal activation of the mammalian target of the rapamycin (mTOR) signaling pathway, which regulates cell growth, proliferation, migration, and cell survival. Clinical presentations range from asymptomatic to dyspnea, cough, hemoptysis, recurrent pneumothoraces, chylous effusions, and respiratory failure. Thus, LAM is sometimes misdiagnosed as asthma or chronic obstructive pulmonary disease.
On HRCT, LAM cysts are typically round and small (usually 2 to 5 mm but as large as 30 mm), without zonal predominance. Small centrilobular nodules in the upper lobe are seen in patients with TSC-LAM, corresponding to micronodular pneumocyte hyperplasia [8]. A characteristic HRCT feature is the presence of > 10 thinwalled, round cysts with preserved or increased lung volume (Fig. 1A), whereas a compatible CT finding is the detection of fewer (> 2 and ≤ 10) cysts [22]. Lymphatic obstruction leads to septal thickening or chylous effusion (Fig. 1B). Pathologically, abnormal smooth muscle-like LAM cells proliferate and result in cystic changes in the lungs (Fig. 1C-1E) and axial lymphatics and angiomyolipomas in the kidney or liver [17]. Although the origin of LAM cells is unknown, the pelvis, especially the uterus, is presumed to be the source because axial lymphatic abnormalities in the pelvis are most common and those distant from the pelvis less frequent. Moreover, LAM cells in the lung are also positive for estrogen/progesterone receptor expression, similar to uterine cells [25,26]. LAM cells may metastasize and recur in the transplanted lung. Like cancer cells, LAM cells depend on glycolysis for energy production; thus, LAM has been defined as a type of “perivascular epithelioid cell tumor” (PEComas) [23].

Lymphangioleiomyomatosis. (A) High-resolution computed tomography (HRCT) shows numerous thin-walled, round cysts scattered in both lung fields with preserved or increased lung volume, suggesting a characteristic HRCT feature of lymphangioleiomyomatosis (LAM). (B) Chylous pleural effusion due to lymphatic obstruction. (C) Gross finding of dilated cystic lesions from surgical biopsy. (D) A low power field of lung tissue shows parenchymal cystic spaces divided by normal lung parenchyma (H&E, ×40). (E) A subpopulation of LAM cells present human melanoma black-45 with intracytoplasmic granular pattern (×100).
The diagnosis of LAM is established by evaluating the HRCT, lung biopsy, and clinical history/findings. According to the European Society Guidelines, a definite, probable, or possible diagnosis is determined after assessing the presence of several requirements [22].
Aside from radiological and pathologic elements, a specific increase in VEGF-D expression (> 800 pg/mL) strongly suggests LAM [27,28]. Typical cystic findings indicative of LAM can be identified on CT, without invasive procedures such as lung biopsy, if more than one of the following are present: TSC, angiomyolipoma, chylothorax, lymphatic involvement, or elevated serum VEGF-D [23].
The management of LAM includes treatment of the pulmonary lesion and control of the concomitant problems. Airflow obstruction is relieved using bronchodila-tors. Recurrent pneumothorax can often be controlled with pleurodesis. Renal angiomyolipomas > 4 cm have an increased risk of bleeding and should be treated with embolization, nephron-sparing surgery, or an mTOR inhibitor [20,22,29]. Sirolimus, an mTOR antagonist, has been associated with improvements in patients with a forced expiratory volume in 1 second (FEV1) < 70%. In patients treated with sirolimus, stable lung function, a better quality of life, improved functional performance, and reduced chylous effusion have been reported [30,31]. However, the optimal dosage of sirolimus, the duration of administration, and the associated side effects have yet to be determined and additional investigation is needed. Everolimus, another mTOR antagonist, was shown to decrease the size of angiomyolipoma and stabilize some lung functions relative to baseline, but its safety issues are similar to those of sirolimus [32,33]. Other potential therapeutic targets include autophagy inhibitors (e.g., chloroquine), MMP inhibitors (e.g., doxycycline), hydroxymethylglutaryl coenzyme A reductase inhibitor (e.g., simvastatin), aromatase inhibitor (e.g., letrozole), cyclooxygenase inhibitor (e.g., aspirin and celecoxib), and tyrosine kinase inhibitors (e.g., axitinib) [20,34,35]. Finally, lung transplantation is an effective option for patients with advanced conditions.
Pulmonary Langerhans cell histiocytosis
This is another rare disease that is predominantly identified in young adults (typically < 40 years of age) but also in cigarette smokers, without a sexual predilection. The clinical presentation of PLCH varies, from asymptomatic to shortness of breath, coughing, weight loss, fever, pneumothorax (15%), skin rash, and diabetes insipidus [36]. Although the exact pathogenesis is unknown, a causal relationship between PLCH and cigarette smoking has been suggested [37-39], as cigarette smoke induces the recruitment and activation of Langerhans cells in the lungs. Langerhans cells are an epithelial-associated dendritic cell and a type of antigen-presenting cell. Cigarette smoke also stimulates the release of several cytokines, such as tumor necrosis factor (TNF-α), granulocyte/macrophage colony-stimulating factor (GM-CSF), and transforming growth factor (TGF-β), which are essential for developing and activating Langerhans cells [40]. The clustering of Langerhans cells and other immune cells around small airways leads to the formation of nodules, airway remodeling, and cystic changes of the lung [4]. Whether PLCH is a clonal proliferative disease, and thus neoplasm-like, or a reactive polyclonal process induced by cigarette smoke is still debated [21,39,41]. However, Langerhans cells undergo clonal proliferation following the mutation of v-Raf murine sarcoma viral oncogene homolog B (BRAF), v-Raf murine sarcoma 3611 viral oncogene homolog (ARAF), mitogen-activated protein kinase kinase 1 (MAP2K1), and other oncogenes, which suggests that PLCH is a neoplastic disorder [21,41].
A pulmonary function test (PFT) in PLCH can indicate findings ranging from normal to restrictive impairment to complete obstruction; therefore, for PLCH patients a PFT is recommended every 3 to 6 months, especially in the first 2 years after diagnosis [4,42].
The most common CT findings in PLCH are cysts and nodules, predominantly in the upper and middle lobes, with almost complete sparing of the costophrenic angles (Fig. 2A). The cysts are often bizarre, in contrast to the round-shaped cysts in LAM or BHD. As the disease progresses, nodules gradually lead to cavitation. The cavitary nodules then become thick/thin-walled cysts and finally confluent cysts [17,43]. If a highly suggestive HRCT finding is made in young smokers, the presence of CD1a-reactive cells > 5% in bronchoalveolar lavage fluid confirms PLCH without biopsy. Otherwise, a surgical lung biopsy is needed for a definite diagnosis. Langerhans cells on light microscopy have an eosinophilic cytoplasm with grooved or convoluted shaped nuclei. These are positive for CD1a and S-100 on immunohistochemical staining. The accumulation of Langerhans and other inflammatory cells around bronchioles causes nodules that are usually as small as 1 to 3 mm in diameter but which may be as large as 2 cm (Fig. 2B-2D). Central cavitation is frequently seen and fibrotic foci are made up of paucicellular stellate scars [9].
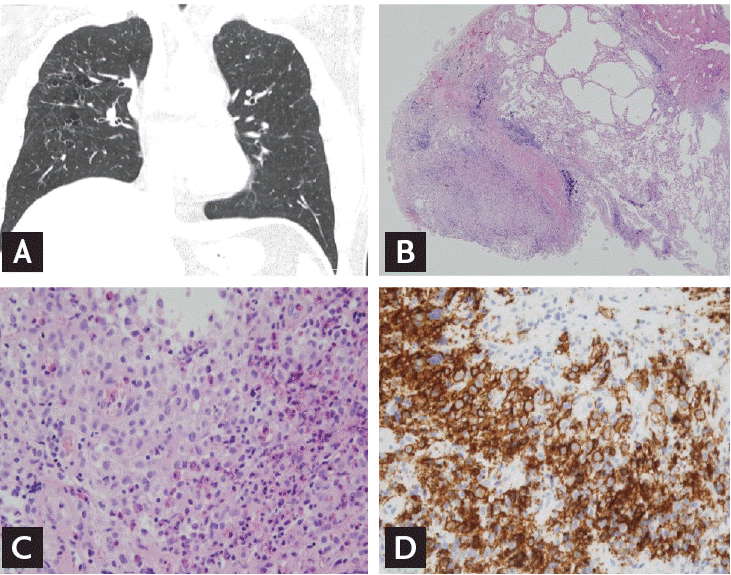
Pulmonary Langerhans cell histiocytosis. (A) High-resolution computed tomography shows multiple bizarre-shaped cysts, especially upper and middle lobe predominance, with almost complete sparing of the costophrenic angles. (B, C, D) Pathologic findings. Histologic features include accumulation of Langerhans and other immune cells around bronchioles (B: H&E, ×40; C: H&E, ×100). Langerhans cells strongly express CD1a on immunohistochemical stain (D: ×100).
Due to the rareness of PLCH, treatment is not standardized, although smoking cessation can be a primary intervention. In some patients, quitting smoking leads to complete PLCH remission, but some patients may recover spontaneously. However, other patients experience progression, including impaired lung function, and should be treated with pharmacotherapy. A corticosteroid is generally used as a first-line drug for PLCH, even though its efficacy is not proven. Other chemotherapeutic agents, including vinblastine, azathioprine, cyclophosphamide, etoposide, methotrexate, and cladribine, have been used in patients unresponsive to corticosteroid therapy [43,44]. With the discovery of targetable mutations such as BRAF, vemurafenib therapy was introduced in patients with BRAF V600 mutations. One study of vemurafenib therapy obtained promising data that indicated a response rate of 43% and disease regression in the majority of patients [45]. Lung transplantation should also be considered in patients with advanced disease. The management of complications is another important issue in patients with PLCH. Pneumothorax that results from cyst rupture is generally managed with pleurodesis because recurrence is common. For pulmonary arterial hypertension, a vasodilator, e.g., endothelin receptor antagonist, phosphodiesterase inhibitor, or prostanoid, may yield clinical improvement without significant worsening of the oxygenation status or pulmonary edema [46].
Brit-Hogg-Dube syndrome
This rare disease has an autosomal dominant inheritance pattern and involves multiple areas of the body, including hair follicle tumors, renal neoplasm, and pulmonary cysts. BHD syndrome is seen in patients in their fourth and fifth decades of life, without a difference in males versus females. Pneumothorax is (typically) recurrent in 75% of BHD syndrome patients. As noted above, the pathogenesis of the disease is a genetic mutation of FLCN, leading to a mTOR signaling abnormality, although whether this mutation causes the activation or inactivation of mTOR is unclear. Another potential pathway is via the abnormal expression of TGF-β or neoplasia (differentially expressed in normal and neoplastic cells [DENN]) protein. FLCN is located on chromosome 17p, and > 140 FLCN DNA mutations have been identified to date (Fig. 3A) [19,47].

Brit-Hogg-Dube syndrome. (A) Folliculin (FLCN) gene analysis showing a missense mutation, which is a substitution of T in exon 4 on chromosome 17. (B) On high-resolution computed tomography, cysts present as multiple thin-walled round, or lentiform, especially distributed in the basilar, medial, and subpleural areas. (C, D) Multiple, small, dome-shaped papules are present on the cheeks with pathologic finding, suggestive of angiofibroma on H&E staining (×100). Adapted from Seo et al. [49].
On HRCT, the cysts in BHD syndrome are multiple, thin-walled, round or lentiform, and well defined. Their size varies from as small as 2 mm to as large as 78 mm [48]. These cysts lack an internal structure and typically have a basilar, medial, and subpleural distribution. The lung volume is commonly preserved or increased, but no other significant pulmonary involvement (i.e., interstitial-lung-disease-like manifestation) has been described (Fig. 3B) [47]. Microscopic findings of the lung tissue in BHD syndrome are generally nonspecific: intraparenchymal air-filled cysts are bounded by normal lung tissue that lacks significant inflammation, an abnormal cellular infiltration, or proliferation. The cystic structure is well maintained by epithelial cells that express surfactant proteins or thyroid transcription factor 1 (TTF-1) [9,15]. The cysts sometimes have an intracystic septum, with an “alveoli within an alveolus” appearance [47].
Along with lung manifestations, potential skin and renal involvement should be meticulously examined. The triad of skin lesion includes fibrofolliculomas, trichodisomas, and acrochordons; the latter are usually multiple, whitish, and dome-shaped papules on the face, neck, or upper back (Fig. 3C and 3D) [49]. The renal pathology can be diverse, ranging from a simple cyst to a malignant tumor. Renal tumors are seven times more common in BHD syndrome patients than in the general population and tend to be bilateral and multifocal [50]. The proposed criteria for a definite/probable/possible diagnosis of BHD syndrome employ CT findings, skin or renal biopsies as well as clinical presentation, as discussed by Gupta et al. [47]. BHD syndrome can be diagnosed in a patient with typical HRCT findings, characteristic skin lesions and/or renal abnormality, a family history, and/or the presence of a related genetic mutation.
The exact clinical course of BHD syndrome has not yet been elucidated, and there is limited knowledge regarding disease progression. Pulmonary function is relatively preserved, but there may be a mild reduction in the diffusing capacity for carbon monoxide. To date, there is no specific treatment for pulmonary cysts; the role of mTOR in this CLD is also unclear [18]. Pleurodesis is recommended for highly recurrent pneumothorax. For the early detection of renal tumors, screening should begin in the patient’s third decade of life and repeated every 3 years. Even though most tumors are relatively indolent, if a tumor > 3 cm is detected nephron-sparing surgical resection should be attempted to reduce the risk of metastasis [19,51].
Lymphocytic interstitial pneumonia/follicular bronchiolitis
LIP is an extremely rare disorder with diffuse involvement of the lung parenchyma by reactive lymphoid tissues, whereas FB is characterized by a lymphoid follicular hyperplasia centered on the airway, vessels, and interlobular septa and presenting as a lymphatic distribution but lacking extensive alveolar septal infiltration [19,52]. The lymphatic distribution in the lung parenchyma may be idiopathic or a secondary change. For instance, Sjögren’s syndrome, rheumatoid arthritis, systemic lupus erythematous, or human immunodeficiency virus infection can present as a lymphocytic congestion in the lung [19]. LIP/FB usually affects females between the ages of 40 and 70 years. Respiratory symptoms, including cough and progressive dyspnea, are frequently present [52].
On HRCT, the cysts in LID or FB are randomly distributed, have an internal structure, measure < 30 mm in diameter, and are typically fewer than in LAM. They frequently accompany GGOs, centrilobular nodules, or septal thickening [36,53]. The PFT of patients with LIP shows a restrictive pattern, whereas in patients with FB the pattern is obstructive. The natural history of LID or FB remains unclear, and the efficacy of immunosuppression, including glucocorticoid as treatment, has yet to be confirmed [19]. While GGOs might be effectively treated with corticosteroid, cystic lesions may be refractory to the immunosuppression, including that by corticosteroids [54].
Amyloidosis
Amyloidosis is an extremely rare and heterogeneous group of disorders characterized by the deposition of a specific protein (amyloid) in extracellular spaces in an abnormal fibrillary fashion. Protein deposition is also noted in hematologic disorders, e.g., multiple myeloma and mucosa-associated lymphoid tissue lymphoma, as well as in autoimmune diseases such as Sjögren’s syndrome [55,56]. A cystic manifestation of pulmonary amyloidosis is rare. CT findings include multiple nodules that may be cavitated (4 to 45 mm in diameter) or calcified (> 1 cm). The cysts are usually multiple, round, and have thin walls (< 2 mm), with a small to moderate size, up to 1 to 2 cm [19,57]. The typical apple-green birefringence on Congo red staining originates from the fibrillary deposits in the lung. There is currently no established curative therapy [18].
CONCLUSIONS
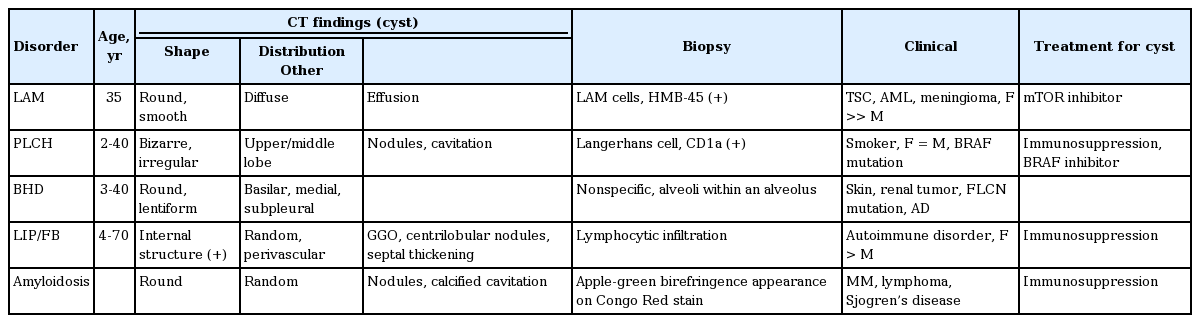
With the increased use of CT, the recognition of CLD has also increased. HRCT is the most valuable diagnostic modality for an initial evaluation of CLD. Along with the characteristic findings of cysts on CT, the pathologic features, presence of genetic mutation, and concomitant disease may be helpful in establishing the differential diagnosis. Table 2 summarizes the diagnostic clues and drug treatment options for each CLD. However, as CLDs are rare, treatment is currently insufficient. Therefore, enhanced knowledge of the pathogenesis of CLDs is needed to guide therapeutic decision-making for each entity.
Characteristics of cystic lung diseases
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
We would like to thank professor Jin Woo Song, from the Asan Medical Center, Seoul, for providing a rare case of BHD for analysis.