Breakthrough in heart failure with preserved ejection fraction: are we there yet?
Article information
Abstract
Heart failure with preserved ejection fraction (HFPEF) is a global health problem of considerable socioeconomic burden. It is projected to worsen with the aging population worldwide. The lack of effective therapies underscores our incomplete understanding of this complex heterogeneous syndrome. A novel paradigm has recently emerged, in which central roles are ascribed to systemic inflammation and generalized endothelial dysfunction in the pathophysiology of HFPEF. In this review, we discuss the role of the endothelium in cardiovascular homeostasis and how deranged endothelial-related signaling pathways contribute to the development of HFPEF. We also review the novel therapies in various stages of research and development that target different components of this signaling pathway.
INTRODUCTION
Heart failure with preserved ejection fraction (HFPEF), a clinical syndrome where heart failure (HF) exists in the presence of normal or near-normal left ventricular ejection fraction (LVEF), accounts for up to half of all HF patients. However, in contrast to heart failure with reduced ejection fraction (HFREF), where therapeutic advances have significantly improved outcomes over the last two decades, there has been little progress in the development of effective, evidence-based therapies for HFPEF. Guideline recommendations for the management of HFPEF have been unchanged for a decade and empirical, largely confined to the use of diuretics and treatment of comorbidities [1,2]. Disappointing results from large phase III HFPEF clinical trials, including the Irbesartan in Heart Failure with Preserved Ejection Fraction Study (I-PRESERVE) [3], Perindopril for Elderly People with Chronic Heart Failure (PEP-CHF) [4], Candersartan in Heart Failure-Assessment of Reduction in Mortality and Morbidity-Preserved Study (CHARM-Preserved) [5], and Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) [6], underscore our incomplete understanding of this disease. At the same time, HFPEF is becoming the predominant form of HF in aging societies [7], and represents a huge public health burden worldwide. Clearly, a break-through is urgently needed to address what is increasingly recognized as the largest unmet need in cardiology today—HFPEF.
BREAKTHROUGH CONCEPT IN HFPEF
A novel HFPEF paradigm emerges from recent studies
A lack of myocardial tissue obtained from HFPEF patients has hampered research in HFPEF pathophysiology for decades, partly accounting for the slow progress in HFPEF research. This is compounded by the absence of truly representative experimental models of HFPEF, given the heterogeneity of the condition. Over the last decade, investigators have obtained myocardial specimens from HFPEF patients, either through endomyocardial biopsies or autopsies. The studies conducted provided important insights into the structural and functional abnormalities seen in HFPEF. The investigators found remodeling involving both the myocardium and extracellular matrix. There was significant hypertrophy of the cardiomyocytes and interstitial fibrosis [8-10]. On a functional level, this translated into increased myocardial stiffness [11] and incomplete myocardial relaxation [12]. In addition, there was evidence of increased inflammation and oxidative stress. Westermann and colleagues [10] showed the presence of profibrotic inflammatory cells in the myocardium of HFPEF patients; this was accompanied by an accumulation of cardiac collagen and correlated with the extent of diastolic dysfunction. Another group of investigators found increased nitrotyrosine content in the myocardium of HFPEF patients compared with patients with HFREF and aortic stenosis; this was accompanied by lower cyclic guanosine monophosphate (cGMP) activity and higher myocardial resting tension, suggesting reduced nitric oxide (NO) bioavailability in an altered redox state [9].
Based on these data on myocardial remodeling and dysfunction, as well as increasing awareness of the important role of comorbidities in the pathophysiology of HFPEF [13,14], a novel HFPEF paradigm was proposed [15]: comorbidities lead to systemic inflammation, increased oxidative stress and generalized endothelial dysfunction. At the level of the myocardium, there is coronary microvascular endothelial inflammation and dysfunction, as well as dysregulation of cardiac endothelium-cardiomyocyte signaling, eventually culminating in the structural and functional alterations seen in the cardiomyocytes and extracellular matrix. Beyond the myocardium, endothelial dysfunction underlies the vasomotor dysfunction seen in various circulatory beds, leading to abnormal ventricular-vascular coupling, pulmonary hypertension, renal dysfunction, and exercise intolerance (Fig. 1) [16]. This novel paradigm not only ascribed a central role to the endothelium in the pathophysiology of HFPEF, but supports the notion that HFPEF is indeed a systemic disorder.
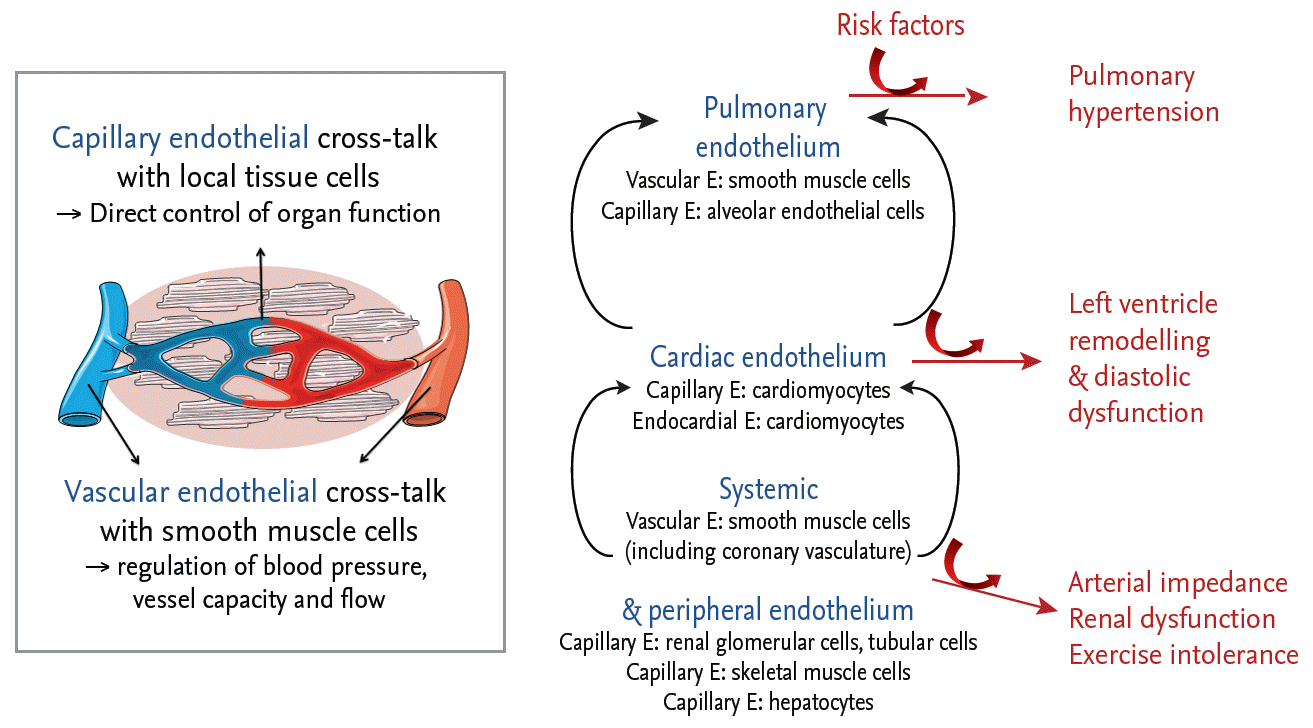
Central role of the endothelium in the pathophysiology of heart failure with preserved ejection fraction. The endothelium plays an obligatory role in cardiovascular homeostasis. While the vascular endothelium plays an important role in the regulation of vasomotor tone, the capillary endothelial cells, which form the largest number of endothelial cells in the body, communicates closely with adjacent organ-specific cells. Proinflammatory risk factors induce generalized endothelial dysfunction, leading to myocardial dysfunction, systemic hypertension, pulmonary hypertension, renal dysfunction, and exercise intolerance. Adapted from Lim et al. [16], with permission from Oxford University Press.
The endothelium plays an important role in cardiovascular homeostasis
The endothelium plays an obligatory role in cardiovascular homeostasis. Endothelial cells regulate vascular permeability, adjust vasomotor tone, maintain blood fluidity, and control inflammatory processes via the expression, activation and release of numerous bioactive factors. The regulatory effects of the endothelium extend beyond the vasculature to include the regulation of myocardial structure and function: endothelial cells modulate inotropy, lusitropy, and chronotropy via their interaction with cardiomyocytes [17]. Numerous endothelial-derived factors have been identified to date, with NO being the prototype of endothelial-derived relaxing factor.
The main signaling pathway of NO is through the activation of soluble guanylate cyclase (sGC), which, in turn, produces cGMP, a downstream mediator. The production of cGMP is not confined to this pathway and is also regulated by the natriuretic peptide (NP)-particulate guanylate cyclase (pGC)-cGMP pathway. This results in the production of two spatially and functionally distinct cGMP pools [18,19]. Intracellular cGMP, in turn, activates protein kinases (protein kinase A [PKA] and protein kinase G [PKG]) and gated ion channels, and regulates phosphodiesterases. The physiologic actions of cGMP differ depending on the site of activation. The activity of cGMP is terminated by the degradation of GMP, catalyzed primarily by phosphodiesterase 5 (PDE-5).
In the vasculature, NO-sGC-cGMP signaling results in the phosphorylation of various membrane proteins residing in the sarcoplasmic reticulum, including phospholamban [20], 1,4,5-inositol triphosphate (IP3) receptor-associated cGMP kinase substrate (IRAG) [21] and calcium (Ca2+)-dependent potassium (K+) channels [22]. This results in reduced intracellular Ca2+ concentration through sequestration of intracellular Ca2+ and reduced influx of extracellular Ca2+ into the sarcoplasmic reticulum. Consequently, there is decreased formation of the Ca2+-calmodulin myosin light chain kinase complex, favoring vasodilatation [23,24]. In addition, NO controls the cellular milieu within the vessel wall; by regulating the synthesis, expression and activity of cytokines, adhesion molecules and growth factors, it modulates platelet aggregation [25], inflammation [26], and smooth muscle proliferation [27].
The effect of NO-sGC-cGMP signaling on cardiac inotropy is bimodal [28,29]. At low and physiological concentrations, cGMP inhibits the activity of PDE III and promotes the intracellular accumulation of cyclic adenosine monophosphate. This, is turn, activates PKA, leading to the opening of sarcoplasmic ryanodine receptors (Ry/R) and sarcolemmal voltage-operated Ca2+ channels, increased intracellular Ca2+ concentration and improved inotropy [30]. At higher concentrations, cGMP improves lusitropy mainly via the activation of PKA and PKG, with phosphorylation of downstream mediator proteins, including troponin I and titin. Phosphorylation of troponin I by PKG reduces cardiac myofilaments’ sensitivity to Ca2+ and promotes diastolic cross-bridge detachment, leading to a rightward shift in the length-tension relationship of the cardiomyocytes [31]. Similarly, phosphorylation of the cytoskeletal protein titin by PKA and PKG its compliance; thus, attenuating myocardial stiffness and improving lusitropy myocardial stiffness and improving lusitropy [32].
NO regulates mitochondrial respiration, promotes free fatty acids as the preferred energy substrate and protects against excessive oxygen consumption. These effects on myocardial energetics complement its effects on lusitropy by limiting myocardial energy wastage in late systole due to the contraction against reflected arterial pressure waves [33].
Endothelial dysfunction plays a central role in the pathophysiology of HFPEF
Cardiovascular risk factors and noncardiac comorbidities are prevalent in HFPEF [34], and induce a systemic proinflammatory state. Indeed, circulating levels of inflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin 6 (IL-6) [35], pentraxin 3 [36], and ST 2 [37] are higher in HFPEF patients. In the myocardium, this leads to the upregulation of coronary microvascular endothelial adhesion molecules, migration of activated circulating leukocytes and increased production of reactive oxygen species (ROS) [15]. The resultant oxidative stress causes endothelial inflammation and dysfunction, impaired NO-sGC-cGMP signaling and reduced activity of downstream mediator proteins PKG and PKA. Consequently, there is an increase in intracellular diastolic Ca2+, enhanced sensitivity of troponin C to Ca2+ and hypophosphorylation of titin. This affects myocardial lusitropy, with delayed myocardial relaxation and increased myocardial stiffness. This is supported by rodent studies that demonstrated a leftward shift of the left ventricle (LV) pressure-volume relationship and worsened LV diastolic function when NO production was inhibited [38]. PKG is also believed to regulate prohypertrophic stimuli and limit cardiomyocyte size, an observation supported by numerous clinical and animal studies [9,39,40]. The final product of a deficient NO-sGC-cGMP pathway is a concentrically remodeled LV with diastolic dysfunction.
The composition of the myocardial interstitial matrix is likewise altered in HFPEF, with increased deposition of type I collagen and greater collagen cross-linking [41]. Monocytes migrating through the inflamed endothelium secrete transforming growth factor β, which promotes the differentiation of fibroblasts into myofibroblasts [10]. This is further aided by the growth-promoting effects of angiotensin II, aldosterone [42], and endothelin-1, whose actions are now unopposed due to low NO bioavailability [39].
Beyond the myocardium, endothelial dysfunction and deficient NO-sGC-cGMP signaling underlie vasomotor dysfunction in various circulatory beds—systemic, pulmonary, renal, and muscular. NO deficiency promotes systemic vasoconstriction and increases arterial impedance, leading to increased LV afterload and abnormal ventricular-vascular coupling.
Pulmonary hypertension is highly prevalent in HFPEF [43] and correlates with peripheral endothelial dysfunction [44], supporting the hypothesis that pulmonary vascular endothelial dysfunction leads to pulmonary vasoconstriction, increased pulmonary vascular resistance and elevated pulmonary artery pressures with eventual right ventricular failure [45].
Renal dysfunction is also highly prevalent in HFPEF, and impaired NO-sGC-cGMP signaling is thought to underlie renal vasomotor dysfunction and cardiorenal syndrome: NO modulates renal glomerular and medullary blood flow, tubular transport, and tubuloglomerular feedback. The activation of sGC in rats with chronic kidney disease improved renal function (as measured by creatinine clearance) and prevented fibrosis (both in the target organ and interstitium) [46]. In a canine model of HF, stimulation of sGC enzyme not only improved cardiac function (increased cardiac output, reduced mean capillary wedge and pulmonary arterial pressures), but also maintained renal function (assessed by glomerular filtration rate) without activation of the renin-angiotensin-aldosterone system [47].
Endothelial dysfunction involving both the resistance and conductive vessels, as well as the microvasculature in skeletal muscles may underlie exercise intolerance, a hallmark of HFPEF. Indeed, Borlaug and colleagues [48] showed that peripheral endothelial dysfunction and impaired exercise-induced augmentation in peripheral blood flow occurred in conjunction with exercise intolerance in HFPEF patients. In addition, microvascular rarefaction is present in the thigh muscles of HFPEF patients, where the capillary density is approximately half that of controls, and this correlated with poorer exercise capacity [49]. It is worth noting that endothelial dysfunction may not be the only player in exercise intolerance in HFPEF patients, as chronotropic incompetence and pulmonary hypertension with exercise-induced elevated left ventricular filling pressures are also likely to be contributors to poor exercise capacity in these patients.
Once HFPEF is established, the accompanying proinflammatory state and neurohormonal activation lead to increased production of ROS and further attenuation of endothelial function. This chronic deficiency in NO-sGC-cGMP signaling further propagates the progression of HF.
Evidence of endothelial dysfunction in HFPEF
The presence of endothelial dysfunction in HFPEF has been demonstrated in numerous studies (Table 1). Impaired endothelial function is present in up to 40% of HFPEF patients [48], afflicting both the conduit vessels and microvasculature. Microvascular rarefaction, and consequent endothelial inflammation and dysfunction, have been shown in both the myocardium [50] and thigh muscles [49], the former being inversely correlated with myocardial fibrosis while the latter correlates with exercise intolerance. In a separate study involving 28 HFPEF patients, peripheral endothelial dysfunction was associated with pulmonary hypertension [44]. Beyond these, endothelial dysfunction is an adverse prognostic marker, independently predicting future cardiovascular events [51,52].
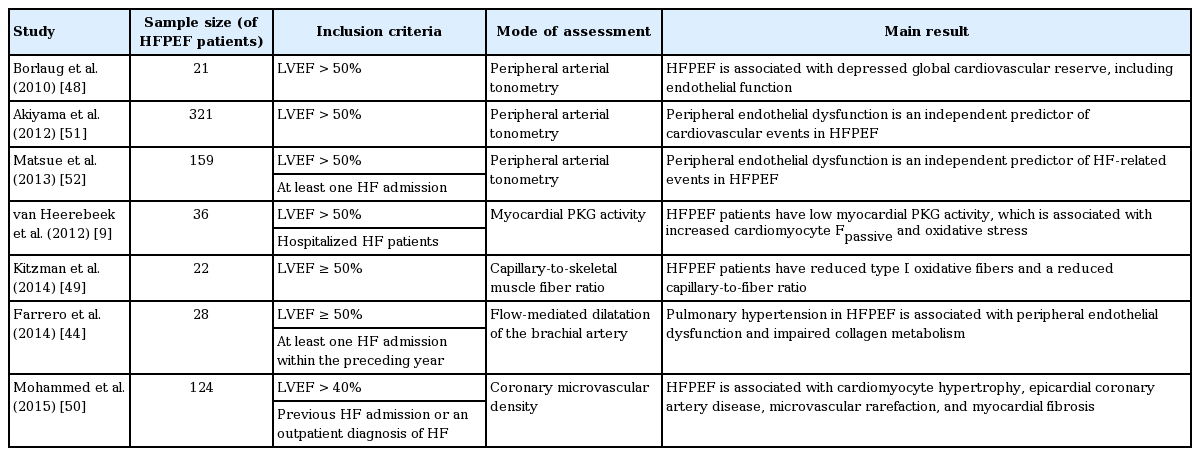
Endothelial dysfunction in patients with heart failure with preserved ejection fraction
HFPEF is a systemic disorder, with comorbidities playing important roles
HFPEF patients are characterized by advanced age and a myriad of cardiometabolic and extracardiac comorbidities, including hypertension, diabetes mellitus (DM), obesity, anemia, chronic lung disease, and chronic renal disease. These comorbidities are generally proinflammatory, and evidence shows both their association with HFPEF as well as endothelial dysfunction.
While the association between DM and atherosclerotic cardiovascular disease has long been established, it is only in recent years that the association between DM and nonischemic HF has gained prominence. Regardless of LVEF, there is a high prevalence of diastolic dysfunction in diabetics [53]. The increased LV stiffness seen amongst diabetics is predominantly due to increased resting tension, without appreciable cardiomyocyte hypertrophy [54]. This unique form of DM cardiomyopathy is believed by some to represent stage B HFPEF [55]. DM is highly prevalent amongst HFPEF patients and is associated with increased production of ROS and impaired antioxidant defense mechanisms. These lead to vascular inflammation with increased levels of inflammatory markers, including IL-6, vascular cellular adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein. The ensuing endothelial dysfunction and deficient NO-sGC-cGMP signaling is generalized, afflicting the central cardiac endothelium as well as the peripheral endothelium. In support of the hypothesis that deficient NO-sGC-cGMP signaling in DM plays a role in the pathogenesis of HFPEF, van Heerebeek and colleagues [53] demonstrated that administration of PKA improved cardiomyocyte resting tension and diastolic function in diabetic HFPEF patients.
As with DM, a significant proportion of HFPEF patients are obese. In fact, the prevalence of HFPEF is rising in tandem with the obesity epidemic. Central obesity interacts with aging in women, resulting in greater LV stiffness. Obesity results in structural and functional changes in the microvasculature, which are attributable to endothelial dysfunction [56]. Microvascular rarefaction and remodeling have been shown in skeletal muscle circulation of obese rats [57] and humans [58]. Functionally, there is an impaired endothelium-dependent vasodilatory response amongst the skin capillaries and resistance vessels [59-61].
Hypertension is a prominent feature amongst HFPEF patients, with a prevalence of 60% to 88% [4,5,62,63]. Not only is it more commonly seen in HFPEF compared with HFREF, but HFPEF patients also have more severe hypertension. Hypertension increases LV afterload, resulting in LV hypertrophy, abnormal ventricular-vascular coupling, myocardial fibrosis, and arterial stiffness. It is an independent predictor of diastolic dysfunction [64]. A unique form of hypertension, pre-eclampsia, has been linked to subsequent development of HFPEF in women [65]. There is a synergistic interaction between hypertension and DM, conferring additional risk of HF in these patients. Hypertension is also proinflammatory, and endothelial dysfunction in hypertensive patients correlates with markers of inflammation, including TNF-α, IL-6, E-selectin, VCAM-1, intercellular adhesion molecule 1, and C-reactive protein [66,67]. As in obesity, structural and functional changes in the microvasculature of hypertensives are observed [68]. Microvascular rarefaction is observed in various circulatory beds, and an increased wall-to-lumen ratio is seen in resistance vessels [69]. Functionally, the balance is shifted in favor of vasoconstriction.
Cumulatively, evidence strongly supports the link between comorbidities, inflammation and an altered redox state, with subsequent endothelial dysfunction in the pathogenesis of HFPEF.
Ethnic differences in endothelial function exist amongst patients
The impact of comorbidities seems more pronounced in Asians than in Caucasians. The wealth of epidemiological data available on HFPEF comes mainly from the United States and Europe, with limited data available on Asians. Currently, the best available data comparing the clinical characteristics of HFPEF among Asians and Caucasians come from the Acute Decompensated Heart Failure National Registry (ADHERE), in which the United States registry is compared against the International (comprising Asia-Pacific and Latin American countries) registry [70]. HFPEF patients from Asia-Pacific and Latin America tend to be younger and have a lower comorbidity burden. Yet, they tend to have more severe clinical presentations, with a longer length of hospitalization and higher rates of mechanical ventilation, the use of inotropic agents and cardiopulmonary resuscitation.
One plausible explanation for these observations is the presence of ethnic differences in endothelial function. Endothelial function is lower in Asians than in Caucasians, regardless of the birth country, implying the presence of genetic susceptibility rather than pure environmental influence [71]. Ethnic differences in endothelial function have similarly been demonstrated in HF patients: Shantsila and colleagues [72] showed that South Asians with HFREF have the worst microvascular endothelial function compared with Caucasians and African Caribbeans with HFPEF.
THERAPEUTIC IMPLICATIONS
The aging population worldwide and rising prevalence of cardiovascular risk factors in Asia and Africa are fueling the HF epidemic, particularly HFPEF. This worsening epidemic, coupled with the lack of evidence-based therapies for HFPEF underscore the urgency of the situation. The emergence of this new paradigm has led to numerous studies evaluating therapies targeting various levels of the NO-sGC-cGMP pathway, and opened up the possibility of novel therapies that may indirectly influence this pathway. Some of these therapies have been tested in clinical trials, whereas others are ongoing.
Recent neutral trials
Sildenafil belongs to a group of drugs known as PDE-5 inhibitors, which enhance intracellular cGMP concentration and activity, thereby improving endothelial function [73]. PDE-5 inhibitors are selective pulmonary vasodilators that are indicated for the management of pulmonary arterial hypertension. In recent years, sildenafil has been explored for use in the management of secondary pulmonary hypertension secondary to chronic HF. Guazzi and colleagues [74] randomized 44 patients with HFPEF and pulmonary hypertension into the placebo or sildenafil arm. Following 1 year of therapy, patients randomized to the sildenafil arm showed improvements in LV filling pressures and pulmonary pressures; these were accompanied by improvements in right and left ventricular function. Findings were less positive when the use of sildenafil was extended to a phase III, multicenter study, the Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure with Preserved Ejection Fraction (RELAX) trial [75]. There was no significant improvement in exercise tolerance, quality of life, hemodynamics, or clinical status after 24 weeks of treatment with sildenafil. A new, long-acting PDE-5 inhibitor, udenafil, is being tested in a 12-week clinical trial (Udenafil Therapy to Improve Symptomatology, Exercise Tolerance and Hemodynamics in Patients with Heart Failure with Preserved Ejection Fraction: Phase III, Randomized, Double Blind, Placebo-controlled Trial [ULTIMATE-HFpEF Trial], ClinicalTrials.gov Identifier: NCT01599117).
Another group of drugs that was thought to improve endothelial function yet yielded neutral findings in clinical trials is the mineralocorticoid antagonists (MRAs). It is well-known that activation of the renin-angiotensin-aldosterone system promotes sodium retention and fibrosis of the myocardium and vasculature. It is now recognized that aldosterone also contributes to endothelial dysfunction. Aldosterone reduces the vasodilatory response to acetylcholine in animal and human studies [76,77], and endothelium-dependent vasodilatation is restored following administration of MRAs [78-80]. Aldosterone contributes to endothelial dysfunction by reducing NO production and increasing oxidative stress. One small study involving 44 HFPEF patients suggested that MRAs may benefit these patients [81].
The Aldosterone receptor Blockade in Diastolic Heart Failure (ALDO-HF) trial was a multicenter, randomized, double-blind trial that randomized 422 HFPEF patients into the spironolactone or placebo arm [82]. At the end of 12 months, spironolactone improved echocardiographic indices of LV diastolic function but failed to improve exercise tolerance and quality of life.
A larger study, the TOPCAT trial, compared spironolactone versus placebo in 3,445 HFPEF patients regarding their clinical outcomes [6]. Although this study failed to significantly improve the primary endpoint, a composite outcome of cardiovascular mortality, HF hospitalizations and aborted cardiac arrest, there was a significant reduction in HF hospitalizations in the spironolactone arm, suggesting that spironolactone had some benefits in morbidity reduction. In addition, a post hoc analysis of the data demonstrated striking regional variations in the outcomes, in which spironolactone significantly improved clinical outcomes in Americas but not in individuals from Georgia or Russia [83].
Ongoing studies
Apart from the concluded studies, there are several ongoing trials targeting the pathway at the NO, sGC, and cGMP levels. NO enhances cardiac lusitropy and myocardial energetics in a synergistic fashion [28,33,84]. Infusion of NO donors, such as nitroprusside, has been shown to improve LV diastolic function and peak systolic pressure [85]. The benefits of organic nitrates were largely demonstrated in HFREF patients, with improvements in mortality, HF hospitalizations and LV function [86,87]. A recent study conducted in 17 HFPEF patients demonstrated an improvement in exercise tolerance following a single dose of inorganic nitrate [88]. This was associated with improvements in peak oxygen consumption, exercise-induced augmentation of peripheral blood flow and endothelial function, along with the attenuation of arterial wave reflections. A larger phase II study is currently underway to evaluate the effects of organic nitrates and hydralazine in HFPEF patients (Effect of Organic Nitrates and Hydralazine on Wave Reflections and Left Ventricular Structure and Function in Heart Failure with Preserved Ejection Fraction, Clinical-Trials.gov Identifier: NCT01516346).
There are two isoforms of sGC that differ by the oxidative state of the prosthetic ferrous heme group. A reduced ferrous heme group renders sGC sensitive to NO, allowing for NO-dependent sGC activation. When oxidized, the heme group dissociates from sGC, resulting in a dysfunctional, NO-insensitive enzyme [89]. sGC stimulators activate the reduced, NO-sensitive form of sGC. These compounds overcome the NO-deficient state by mimicking NO. On the other hand, sGC activators work independently of NO bioavailability. They specifically target the NO-insensitive sGC, activating the enzyme by occupying the free sGC heme-binding site. Data from animal studies have demonstrated that both sGC stimulators and activators have potent vasodilatory and blood pressure-lowering effects, in addition to cardio- and reno-protective properties [90-93].
Riociguat was the first oral sGC stimulator studied in HF patients. The Acute Hemodynamic Effects of Riociguat in Patients with Pulmonary Hypertension Associated with Diastolic Heart Failure (DILATE-1) trial was a randomized, placebo-controlled, parallel group IIa study evaluating the effects of riociguat in HFPEF patients with pulmonary hypertension [94]. A single dose of riociguat (2 mg) improved cardiac output, and decreased afterload and right ventricular dimensions. However, there was no significant change in pulmonary pressure or pulmonary vascular resistance. Other phase II studies are ongoing. Part of the Soluble Guanylate Cyclase stimulator Heart Failure Studies (SOCRATES) program (ClinicalTrials.gov Identifier: NCT101951638), SOCRATES-PRESERVED, is a randomized, double-blind, parallel-group study evaluating the pharmacodynamics, pharmacokinetics and safety profile of vericiguat (BAY1021189) in hospitalized HFPEF (LVEF ≥ 45%) patients following initial stabilization.
NPs act in parallel with NO to increase intracellular cGMP levels. The degradation of NPs occurs via two mechanisms—enzymatic breakdown by neprilysin and receptor internalization followed by lysosomal degradation. LCZ696 is a novel molecule that combines the neprilysin inhibitor prodrug AHU377 with the angiotensin II receptor blocker (ARB) valsartan [95]. Inhibition of neprilysin enhances NP-pGC-cGMP signaling, while inhibition of the angiotensin II receptor suppresses the detrimental activation of the renin-angiotensin-aldosterone. The phase II Prospective Comparison of ARNI (angiotensin receptor neprilysin inhibitor) with ARB on Management of Heart Failure with Preserved Ejection Fraction (PARAMOUNT) trial, which compared the LCZ696 against valsartan for 12 weeks in 301 patients, yielded some results [96]. At 12 weeks, there was a significant improvement in the levels of N-terminal prohormone of brain natriuretic peptide in the LCZ 696 group that was not sustained at 36 weeks. Left atrial dimensions and volumes were significantly reduced, albeit without a concomitant improvement in LV diastolic indices, at 36 weeks in the LCZ696 group. A larger phase III trial (Efficacy and Safety of LCZ696 Compared with Valsartan, on Morbidity and Mortality in Heart Failure Patients with Preserved Ejection Fraction [PARAGON-HF], Clinical-Trials.gov Identifier: NCT01920711) is ongoing, in which LCZ696 will be compared with valsartan on clinical end-points in HFPEF.
Promising pharmacological and nonpharmacological interventions
The proposal of this novel HFPEF paradigm where substantial weight is attributed to comorbidities and systemic inflammation has led to novel therapeutic approaches being considered and tested.
Statins possess anti-inflammatory properties and could potentially exert a stabilizing effect on the endothelium. The therapeutic effects of statins are well-established in the management of dyslipidemia and coronary artery disease, but less so in HF. Although statins reduce myocardial stiffness and fibrosis in experimental models and HFPEF patients, available observational studies have yielded mixed findings regarding the mortality effects in this group of patients. A recent meta-analysis involving almost 18,000 HFPEF patients showed a 40% reduction in mortality rate with statin use [97]. Nonetheless, its beneficial effects need to be confirmed in a properly designed randomized clinical trial.
The formation and accumulation of advanced glycation end products (AGEs), through the interaction of carbohydrates and proteins, occur in DM; this contributes to the development of diastolic dysfunction and subsequent HFPEF directly by increasing myocardial stiffness through the cross-linking of collagen, or indirectly by increasing oxidative stress and reducing NO bioavailability [98]. Plasma levels of AGEs predict worse clinical outcomes in HFPEF [99]. AGE cross-link breakers, such as alagebrium (ALT-711), have been shown to improve left ventricular diastolic function and quality of life after 16 weeks of follow-up in one small study [100]. A larger phase II study (The Beginning A Randomized Evaluation of the AGE Breaker Alagebrium in Diastolic Heart Failure [BREAK-DHF 1], ClinicalTrials.gov Identifier: NCT00662116) was designed to evaluate the efficacy of alagebrium on exercise capacity in HFPEF patients; however, it was terminated early due to financial constraints. Presently, another study is underway evaluating the effect of alagebrium on myocardial stiffness in sedentary or active elderly volunteers (The Effect of Exercise and Alagebrium on the Diastolic Function of the Heart [AGE], ClinicalTrials.gov Identifier: NCT01014572).
Exercise intolerance is a hallmark of HFPEF, and it is postulated that exercise training could benefit HFPEF patients through improvements in endothelial function [101]. Exercise increases endothelial shear stress, enhancing NO release and NO-mediated vasodilatation in HF. In addition, it indirectly improves endothelial function and NO bioavailability through its anti-inflammatory effects, in part by influencing circulating levels of insulin, glucose and cytokines. Exercise training has been shown to improve exercise tolerance, quality of life and indices of LV diastolic function in HFPEF patients [102,103]. The benefits were initially assumed to be secondary to improvements in endothelial function. However, a study carried out by Kitzman and colleagues [104] showed no significant change in endothelial function (assessed by brachial artery flow-mediated dilatation), despite improvements in peak oxygen consumption following exercise training. One possible reason is that the duration of exercise training in this study was insufficient to effect changes in endothelial function. Additionally, conduit arterial endothelial function is not synonymous with microvascular endothelial function, and it remains plausible that exercise training positively affects microvascular reserve without obvious changes in conduit arterial endothelial function. Finally, it is noteworthy that exercise intolerance in HFPEF is multifactorial, with contributions from exercise-induced elevations in LV filling pressures and pulmonary pressures, as well as impaired augmentation of the heart rate and stroke volume during exercise in addition to endothelial dysfunction.
Planned weight loss in obese patients, either via life-style modifications or surgical methods, may mitigate obesity-related cardiovascular complications. Reducing excess adiposity reduces inflammation, and favorably influences endothelial function and insulin sensitivity [105,106]. Impairment of macrovascular endothelial function has been shown following weight gain in normal-weight adults; this phenomenon is reversible once the additional weight is shed [107]. Beyond these, weight reduction has also been shown to improve cardiomyocyte hypertrophy and LV diastolic dysfunction in obese individuals [108-110], which may potentially retard the progression to symptomatic HFPEF.
CONCLUSIONS
HFPEF is an important public health problem of high morbidity and mortality. Although significant inroads have been made regarding its pathogenesis, the results from neutral trials remind us of the gaps in our understanding of this heterogeneous syndrome. With HFPEF prevalence increasing worldwide, including the developing nations, the race for effective therapies has never been more urgent.
Notes
CSPL is supported by a Clinician Scientist Award from the National Medical Research Council of Singapore. CSPL has received research support from Boston Scientific, Medtronic and Vifor Pharma, and has consulted for Bayer and Novartis.