Recent technological updates and clinical applications of induced pluripotent stem cells
Article information
Abstract
Induced pluripotent stem cells (iPSCs) were first described in 2006 and have since emerged as a promising cell source for clinical applications. The rapid progression in iPSC technology is still ongoing and directed toward increasing the efficacy of iPSC production and reducing the immunogenic and tumorigenic potential of these cells. Enormous efforts have been made to apply iPSC-based technology in the clinic, for drug screening approaches and cell replacement therapy. Moreover, disease modeling using patient-specific iPSCs continues to expand our knowledge regarding the pathophysiology and prospective treatment of rare disorders. Furthermore, autologous stem cell therapy with patient-specific iPSCs shows great propensity for the minimization of immune reactions and the provision of a limitless supply of cells for transplantation. In this review, we discuss the recent updates in iPSC technology and the use of iPSCs in disease modeling and regenerative medicine.
INTRODUCTION
Stem cells are pluripotent cells with the capacity for self-renewal and differentiation into various types of adult cells [1]. Ever since their discovery in 1960, stem cells have formed the hub of countless explorations of disease, regeneration, and indeed, "the secret of life" itself [2,3]. Stem cells are roughly categorized as embryonic stem cells (ESCs), mesenchymal stem cells (MSCs), and induced pluripotent stem cells (iPSCs). ESCs are derived from the inner cell mass of preimplantation embryos and demonstrate excellent pluripotency, but their use is confounded by ethical issues regarding the destruction of the blastocyst [4]. MSCs are obtained from adult adipose tissue, blood, bone marrow, and cord blood and are thus free from ethical concerns, but their use is still limited by low cell numbers and diminished pluripotency. However, the more recently introduced iPSCs show enormous promise for disease modeling and regenerative medicine because their pluripotency is similar to that of ESCs, while their utilization is without ethical controversy [5].
Mouse iPSCs were first introduced by Takahashi and Yamanaka [6] in 2006 by generating mouse pluripotent stem cells from dermal fibroblasts via transduction with four reprogramming factors, octamer-binding transcription factor 4 (Oct4), Kruppel-like factor 4 (Klf4), sex determining region Y-box 2 (Sox2), and c-Myc (i.e., the canonical Oct4, Sox2, Klf4, and c-Myc [OSKM] quartet). The discovery of iPSCs dramatically altered the previous dogma of cellular differentiation as a unidirectional, nonrevertible developmental process, resulting in a paradigm shift in the field of developmental biology. Furthermore, the work of Takahashi and Yamanaka [6] provided the foundation for an entirely new field of research in cell reprogramming and translational medicine. The newly generated mouse iPSCs were nearly indistinguishable from ESCs, in that iPSCs and ESCs can both proliferate indefinitely under controlled culture conditions and then differentiate in vivo and in vitro into all cell types, making iPSCs and ESCs alike an attractive cell source for translational and regenerative medicine applications. However, ethical concerns, limited availability, and possible immunogenicity are the main disadvantages of ESCs over iPSCs.
Generation of human iPSCs followed the production of mouse iPSCs, and like mouse iPSCs, the human equivalent escapes the ethical conundrum of blastocyst destruction. In addition, self-derived autologous human iPSCs now enable the ready attainment of human leukocyte antigen (HLA)-full matched stem cells without the effort of searching the human HLA bank database. Acquisition of an immunologically tolerant stem cell source will undoubtedly facilitate the future utilization of iPSCs in the field of human regenerative medicine. Furthermore, patient-specific iPSCs may open a new field of personalized medicine, represented by novel "patient in a dish" and "patient in a tube" explorations [2,7]. Indeed, disease modeling with patient-derived iPSCs has been successfully used to clarify the pathophysiology of several rare and/or incurable diseases, including retinal degeneration, spinal muscular atrophy, and Alzheimer's disease. The next step will be to employ these iPSC-based disease platforms for a thorough molecular analysis of the disease phenotype in question, followed by large-scale drug screening and new drug development for disease management.
In this review, we recapitulate the recent progress made in the area of iPSC technology. In the first part of the review, we summarize recent techniques for iPSC generation (i.e., viral and episomal vector-mediated reprogramming, as well as microRNA [mRNA]- and protein-mediated induction of pluripotency). We also discuss gene editing to correct genetic defects in iPSCs and to produce resultantly sound stem cells. In the second part of the review, we deliberate upon assorted clinical applications of iPSCs, from the standpoint of recent feasibility and future possibilities.
PART 1. RECENT UPDATES IN iPSC GENERATION
In 2006, Takahashi and Yamanaka [6] demonstrated that terminally-differentiated somatic cells can be reverted into a cell type having enhanced developmental potential by overexpressing transcription factors that regulate the maintenance of ESC pluripotency. OSKM were identified as the most important reprogramming factors for the induction of pluripotency following a screening of 24 genes which were virally overexpressed in mouse embryonic fibroblasts [6]. These four factors synergistically activate the molecular circuitry of pluripotency, which converts the differentiated somatic cell into an undifferentiated pluripotent cell [8].
In 2007, Takahashi et al. [9] and Yu et al. [10] successfully reproduced their groundbreaking work with mouse fibroblasts in human fibroblasts. This was accomplished by using either the same combination of factors (OSKM), or human Oct4 and Sox2 together with Nanog and LIN28. Subsequent studies revealed that reprogramming efficiency could be significantly increased by using polycistronic reprogramming constructs, chromatin-modifying chemicals, and mRNAs, as well as through activation or inhibition of various signaling pathways involved in the regulation of cell proliferation [11,12,13,14]. Moreover, Bayart and Cohen-Haguenauer [15] showed that individual reprogramming factors could be exchanged or entirely removed from the reprogramming cocktail without losing the capacity to induce pluripotency in somatic cells.
Conventional reprogramming techniques depend on the stable integration of transgenes but introduce the concurrent risk of insertional mutagenesis [16]. Several nonintegrating reprogramming techniques have thus been developed to circumvent the risk of spontaneous tumor formation and to improve the quality of the generated iPSCs. Some of these techniques are grounded on the almost complete removal of the integrated viral DNA or alternatively, on the use of nonintegrating viruses [17,18]. Furthermore, the launch of virus-independent reprogramming methods based on DNA, protein, or mRNA expression is expected to further improve iPSC quality [19,20,21].
The following sections summarize the recent advances in reprogramming technology for the derivation of iPSCs (including patient-specific iPSCs), as well as gene editing techniques for the generation of modified iPSCs.
Generation of clinically feasible iPSCs: an overview
For the purposes of clinical application of human iPSCs, it is important to choose the correct donor cell type and the best reprogramming method. The perfect donor cell should be easy to obtain, ideally from the patient him or herself, and to reprogram. Therefore, fibroblasts, keratinocytes, and peripheral blood mononuclear cells (PBMCs) are all preferred cell types as the initiation point for the induction of pluripotency. Recent efforts to establish large-scale iPSC biobanks have concentrated their efforts on PBMCs because they can be readily attained via phlebotomy and are robustly reprogrammable [22].
Safety is the major issue surrounding cell generation for translational applications. Therefore, non-integrative reprogramming methods are favored over integrative methods, limiting the possibility of internal mutagenesis and consequent tumor formation. Below, we describe the most frequently used nonintegrative reprogramming techniques and discuss their comparative advantages and drawbacks.
Reprogramming using the Sendai virus
Most iPSC laboratories use F-deficient Sendai virus particles to induce pluripotency in somatic cells. The Sendai virus is a negative-sense, single-stranded RNA virus, and replicates in the cytoplasm of infected cells without DNA intermediates or stable integration into the target cell genome [23]. Sendai virus-mediated reprogramming is perhaps the most efficient integration-free method of iPSC production currently available and was previously used to effectively reprogram fibroblasts and PBMCs [23,24].
However, besides being an expensive method, the major drawback of using the Sendai virus is the persistence of residual viral material. The latter requires an extended period of tissue culture (10 to 20 passages) to establish virus-free iPSC lines for further downstream analysis and differentiation experiments [14]. To overcome this limitation, specific mutations have been introduced into key viral proteins to develop temperature-sensitive Sendai viral particles, allowing for faster and more efficient removal of viral material from the host cell cytoplasm via a temperature shift [25]. Nonetheless, working with Sendai viral particles still requires stringent biosafety containment measures and a separate tissue culture room, further increasing the costs of the procedure.
DNA-based episomal reprogramming
As an alternative to the viral-mediated induction of pluripotency, several DNA-based reprogramming methods have been developed by using either non-replicating [20,26,27,28] or replicating episomal vectors [29]. These techniques are attractive because they reduce the biosafety concerns involved in the production and transduction of viral particles. However, the reprogramming efficiency of non-replicating vectors is rather low, requiring multiple transfections of the target cells [20,26,27]. A possible explanation for this phenomenon is provided by the low transfection efficiency of large polycistronic reprogramming plasmids in addition to the transgene silencing mechanism of plasmid-based vectors in mammalian cells [30]. To overcome these obstacles, Jia and colleagues [20] developed minicircle vectors as a shuttle system for the reprogramming factors. Minicircles consist of a special episomal vector devoid of any bacterial plasmid and are therefore smaller in size than standard plasmids. These properties substantially enhance the transfection efficiency and expression rate of minicircles [31]. However, the reprogramming efficiency of minicircles remains quite low, and the production and purification methodology associated with this vector system is relatively complex and fairly time-consuming [32].
Accordingly, we developed a novel single plasmid reprogramming system termed the codon-optimized 4-in-1 minicircle (CoMiP), which carries codon-optimized sequences of the canonical OKSM reprogramming quartet and a short hairpin RNA against the p53 tumor suppressor [26]. Similar to minicircles, the CoMiP vector system overcomes the transgene silencing observed with regular plasmids and provides at least 2- to 10-fold higher levels of transgene expression [33]. Furthermore, the CoMiP vector is a highly efficient, integration-free, cost-effective agent and is applicable to a wide variety of cell types, including fibroblasts and PBMCs.
Another compelling methodology for the derivation of human iPSCs involves the binding of Epstein-Barr virus-encoded nuclear antigen-1 (EBNA-1) to a cis-acting viral DNA element, oriP. The EBNA-1/oriP association permits the persistence of plasmids in actively dividing human cells as multicopy episomes that attach to chromosomes during mitosis [34,35]. After multiple cell divisions, oriP/EBNA-based vectors are progressively lost from the targeted host cells. Nevertheless, these vector systems significantly increase the reprogramming efficiency through the prolonged expression of transgenes within the transfected cell [35]. However, DNA-based reprogramming methods could potentially lead to the integration of foreign DNA into the host genome and therefore require accurate downstream screening methods to confirm the derivation of integration-free pluripotent cells [16,27].
Induction of pluripotency via mRNA or protein expression
The continuous, transient transfection of modified mRNAs into parental cells is an elegant approach for the induction of pluripotency that guarantees derivation of integration-free iPSCs [21,36]. Because this method is independent of a DNA intermediate, it circumvents prospective integrations and thus, time-consuming screening experiments. However, modified mRNA transfection still requires pre-treatment of target cells with the expensive interferon alpha antagonist, B18R, and a laborious series (up to 14 days) of mRNA transfections [36]. The B18R protein significantly enhances cell survival after the series of transfections by blocking the interferon alpha signaling pathway [37]. Another limitation of mRNA-mediated reprogramming for clinical approaches is the requirement of a feeder cell layer and feeder cell-derived conditioned media, both of which can increase the risk of transmitting undetected human pathogens to the host [38]. The recent optimization of established mRNA reprogramming factors has addressed some of the aforementioned caveats, allowing the production of footprint-free iPSCs from human fibroblasts without the use of feeder cells or other possibly xeno-contaminated reagents [39]. Furthermore, Yoshioka and colleagues [40] successfully generated "clean" iPSCs from newborn or adult human fibroblasts by a single transfection of a synthetic, self-replicative RNA. However, further validation of this approach is necessary to establish its vigor and reproducibility.
Delivery of reprogramming factors to cells as transmembrane permeable fusion proteins is another means of inducing pluripotency that prevents possible alteration of the target cell genome [41,42]. Despite the promise of this strategy, the protein-mediated reprogramming approach is hampered by slow kinetics, inefficiency, low reproducibility, and high cost [16]. Recently, an exciting alternative approach was introduced that exclusively uses small-molecule compounds to reprogram mouse somatic cells [43]. However, the efficiency of this technique is also quite low, and the study must be reproduced by using human rather than mouse somatic cells to achieve broader clinical interest.
Methodologies for the generation of clinically feasible iPSCs are still under development. The successful advance of integration-free techniques for the induction of pluripotency, as well as xeno-free culture conditions both during and after the reprogramming process, would be pivotal for future translational applications.
Targeted editing of the iPSC genome
Traditionally, human ESCs were modified by using laborious and inefficient transfection protocols, homologous recombination, and clonal expansion [44]. Recently, tremendous progress has been made in terms of stem cell transfection, cultivation, and genome editing. Defined culture conditions together with small-molecule reagents and advanced stem cell transfection protocols provided the initial foundation for the development of new editing procedures for pluripotent stem cells. Currently, three main technologies are used to target and correct a wide variety of mutations in iPSCs: the zinc finger nuclease (ZFN) system, the transcription activator-like effector nuclease (TALEN) system, and the clustered regularly interspaced short palindromic repeats (CRISPR) system. Gene editing with ZFNs and TALENs utilize programmable, sequence-specific DNA-binding domains linked to a nonspecific DNA cleavage domain, Fok1, to form a functional dimeric nuclease [45,46]. On the other hand, the CRISPR system takes advantage of the RNA-guided Cas9 nuclease to generate directed double-stranded DNA breaks [47].
Although ZFNs were initially used for genome editing experiments, the TALEN and CRISPR systems are now preferable due to their relatively low expense and ease of assembly. However, which of the three methods has the highest cutting efficiency without additional off-target effects remains to be determined [48,49].
Genome engineering strongly depends on cellular responses to DNA damage. For example, induction of double-strand DNA breaks triggers either error-prone, nonhomologous end joining or homology-directed repair at specific genomic locations, leading to small insertions/deletions at the target site or introduction of a homologous donor DNA template, respectively [49]. Based on these repair mechanisms, it is possible to derive heterozygous/homozygous knockout cell lines or to precisely introduce/correct specific gene mutations. The precise correction or introduction of a particular mutation in the same genetic background allows a more accurate way of disease modeling. Using these so called isogenic cell lines are the foundation to elucidate the underlying molecular mechanism of a certain disease.
In summary, recent advances in integration-free reprogramming technology and genome engineering, combined with the enormous differentiation capacity of pluripotent cells, renders iPSCs an ideal tool for translational research (Fig. 1).
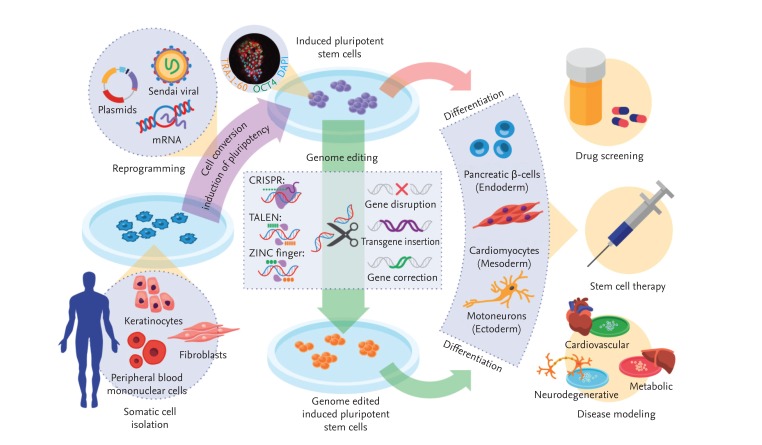
Generation of patient-specific induced pluripotent stem cells (iPSCs) and clinical applications thereof. Somatic cells isolated from a patient are reprogrammed into iPSCs by transduction with the four reprogramming factors, octamer-binding transcription factor 4 (Oct4), sex determining region Y-box 2, Kruppel-like factor 4, and c-Myc. Genetic defects in iPSCs can be corrected via gene editing with zinc finger nucleases (ZFNs), activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeats (CRISPR) system. Next, iPSCs with or without edited modifications are differentiated into various target cells for disease modeling, drug screening, and stem cell therapy. DAPI, 4',6-diamidino-2-phenylindole.
PART 2. CLINICAL APPLICATIONS OF iPSCS
Recent advances in stem cell technology are likely to greatly expedite the clinical use of iPSCs in applications for human patients. These cells are an attractive candidate for research because they can assume the individual characteristics of multiple cell types, including disease-relevant cells. Although ESCs share the proliferative capacity and multipotency of iPSCs, their use in pathophysiological research is limited due to their inability to take on a disease-selective phenotype. By contrast, patient-derived iPSCs retain specific genetic and epigenetic memories of the individual from which they originated, enabling the simulation of the patient's own disease. Currently, disease modeling of several disorders has been realized by using disease-relevant cells differentiated from patient-specific iPSCs. This extraordinary achievement has resulted in the pioneering of a new field of research for the deduction of pathogenic disease mechanisms, as well as for preclinical drug screening.
Therapeutic approaches based on iPSC-mediated cell and tissue transplantation are also promising, given the unlimited proliferative capacity of iPSCs to provide sufficient quantities of cells for stem cell therapy. Moreover, patient-specific iPSCs can hypothetically minimize immune reactions and reduce the risk of graft-versus-host rejection. However, several barriers must be overcome before the successful clinical application of iPSCs. In this part of the review, we discuss the feasibility and concerns of iPSCs in association with disease modeling, drug screening, and regenerative medicine.
Disease modeling and drug screening
Nowadays, disease simulation is a mainstream of iPSC applications in research laboratories and in clinics [2,50,51]. Disease modeling with iPSCs recapitulates a pathologic condition in vitro by reprogramming a patient's somatic cells into iPSCs, followed by redifferentiation of the patient-specific iPSCs into disease-specific cell types [52,53,54,55,56,57,58,59,60]. This approach is favored because risk and harm to the patient are minimal compared with other cell transfer therapies; furthermore, a vast number of target cells can be consistently generated from patient-specific iPSCs with infinite expansion capacity. If relevant expanded iPSCs and target cells were to be globally distributed to researchers and drug development teams, the performance of standardized and directed research in a specific disease area might become possible. This feat would increase not only the safety and feasibility of cell source but also the accuracy of disease simulation.
Many reports have documented discrepancies between mouse disease models and actual human disease [61,62]. Investigations of iPSCs generated from cells and tissues directly harvested from patients would likely minimize the fallacies originating from such discrepancies. Researchers would then be able to simulate the conditions caused by aberrant genes of patient-specific iPSCs in a dish or in a tube. On the other hand, the expense incurred by the generation of certain genetic mutations in cell lines or animal models can be an economical roadblock to large-scale drug development and disease modeling [2,63]. From this point of view, the generation of patient-specific iPSCs often costs less than the generation of animal disease models or genetically engineered animals. Therefore patient-specific iPSC drug platform might be a good candidate for a preclinical validation tool in terms of economic feasibility and patient safety [64]. If some drugs are successful in such an iPSC platform, drug development may then proceed to the next stage with more confidence and less risk. Overall, safety, feasibility, accuracy, and reasonable cost all favor the research and application of patient-specific iPSCs for disease modeling and drug screening.
Nevertheless, disease modeling with iPSCs has several limitations and concerns, including lack of homogeneity in many iPSC cell lines. In vitro disease modeling is affected by cellular artifacts and culture conditions. Thus, if environmental factors readily affect iPSC properties and differentiation into target cells, reproducibility of disease modeling with patient-specific iPSCs becomes a formidable issue. Although target cells are differentiated from stable iPSCs, the former exist in a spectrum, from immature cells to mature and functional cells. Therefore, target cell diversity is indisputably a barrier to the simulation of disease in a dish [4,65].
Second, the complexity of disease pathophysiology may not be revealed by any single cells derived from patient-specific iPSCs, because cell-to-cell interactions also play important roles in intractable diseases. Therefore, simulation of a complex disease requires a more complicated and elaborate system rather than that afforded by isolated cells and cellular components [66]. For this reason, the initial stages of iPSC application to disease modeling have concentrated on straightforward genetic disorders, given that the pathologically autonomous iPSC cells generated in a monogenic disease represent the main phenotype of the that condition [67].
Third, disease modeling faces challenges if the disease pathogenesis itself is influenced by the environment. Host susceptibility and environmental factors critically interact in the progression of chronic degenerative and metabolic conditions. Accordingly, efforts have been made to lessen the gap between disease simulation and reality. For instance, environmentally-induced senescence can be mimicked in disease modeling by the addition of specific molecules to the culture system, such as progerin, which reportedly induces premature aging in stem cells [68]. This type of strategy will be advantageous for accurate simulations of chronic human degenerative diseases.
Regenerative medicine
Stem cells show great promise for the minimization of immune rejection in regenerative medicine, as successfully demonstrated by attempts to alleviate signs and symptoms in disease-relevant animal models. For example, Parkinson's disease model rats were effectively treated by cell replacement therapy with terminally-differentiated neurons derived from reprogrammed fibroblasts, with little immune rejection [69]. Moreover, iPSCs corrected through gene editing displayed the therapeutic potential to cure genetic disorders in a mouse model of sickle cell anemia, together with reduced immunogenicity [70].
Regenerative medicine approaches with iPSCs have several clinical merits. Above all, patient-derived iPSCs are immunologically privileged upon autologous transplantation [71], possibly eliminating the need for lifelong immunosuppressive drugs. Self-renewal and everlasting proliferation are another merit of iPSCs in regenerative medicine. As noted above, the proliferative capacity of iPSCs is equivalent to that of ESCs; hence, iPSCs represent a limitless source of cells for cell replacement therapy. Moreover, the pluripotency of iPSCs enables the formation of a functional organ structure beyond what is capable with a mixture of cells and cellular components [72]. However, the development of new biomaterials and appropriate scaffolds will be necessary to fully support the clinical application of iPSCs in human medicine.
Nonetheless, iPSC-based cell therapy is hampered by substantial obstacles. In particular, the clinical use of these cells requires ongoing and strict guidelines to minimize harmful outcomes to patients while maximizing patient safety. Although technical progress continues, there remains a tremendous gap between iPSC generation for research purposes and for therapeutic purposes.
Good manufacturing practice (GMP) and standard operating procedures (SOP) are important terms that are frequently encountered in the clinical application of stem cells. GMP and SOP principles necessarily escalate the expense of iPSC generation and manipulation. Thus, the balance between cost and safety remains an issue. However, despite this caveat, iPSC therapy seems imperative for certain conditions. For example, neurodegenerative and cardiac diseases have limited treatment options other than regenerative therapy because neurons and cardiomyocytes are not renewable. Stem cell strategies, including the use of iPSCs, are encouraging for the replacement of lost nerve or cardiac tissue and other non-renewable tissues or organs. On a practical note, the first clinical trial of iPSCs was recently approved for the treatment of age-related macular degeneration in Japan [73].
PERSPECTIVE
In 2012, John B. Gurdon and Shinya Yamanaka were awarded the Nobel Prize in Medicine for their pioneering work on cellular plasticity and cellular differentiation, only 6 years after Yamanaka initially generated iPSCs in 2006 [6]. This accomplishment indirectly highlights the intense focus placed on iPSC research over the past several years. Vast amounts of research funding and manpower have been invested in iPSC technology to widen the opportunity for new clinical applications and hasten the realization of stem cell therapy.
In the near future, we anticipate that patient-specific iPSCs will prove increasingly useful for in vitro disease modeling. Diseases with an unclear pathogenic mechanism are an obvious target for vigorous pathophysiological research with such patient-derived cells. This approach covers the gamut from large-scale preclinical drug screening with globally available, disease-selective iPSCs to patient-specific, tailored medicine.
Safety and availability are the main concerns for iPSC-based therapy in clinical applications. Standardized manufacturing in a qualified institution or company is the minimum condition for implementing and securing safety standards. In addition, prompt use of iPSCs for tissue or organ transplantation requires that the cells be ready on short notice for universal purposes. For these reasons, national stem cell banking based on HLA expression in stem cells is under government regulation [74,75,76,77].
CONCLUSIONS
In conclusion, the application of iPSCs to clinical research is an ongoing hot topic, although limitations of iPSCs must be taken into account. Patient-specific iPSCs provide crucial new tools for the continuous expansion of our knowledge regarding disease pathogenesis and treatment, and cell therapy with patient-specific iPSCs is advantageous from the viewpoint of immune tolerance and an endless supply of cells. These beneficial stem cell properties will almost assuredly encourage further exploration of iPSCs for disease modeling and regenerative medicine applications, both in the laboratory and in the clinic.
Acknowledgments
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A1A1076125) and Grant for "the Center for Evaluating Next-Generation Stem Cell-based Therapeutics" supported by National Institute of Food and Drug Safety Evaluation, part of the Ministry of Drug and Food Safety (14172 CENST 974), and a grant from German research foundation (DFG) DI1877/1-1.
Notes
No potential conflict of interest relevant to this article was reported.