Impact of Air Pollution on Allergic Diseases
Article information
Abstract
The incidence of allergic diseases in most industrialized countries has increased. Although the exact mechanisms behind this rapid increase in prevalence remain uncertain, a variety of air pollutants have been attracting attention as one causative factor. Epidemiological and toxicological research suggests a causative relationship between air pollution and the increased incidence of asthma, allergic rhinitis, and other allergic disorders. These include ozone, nitrogen dioxide and, especially particulate matter, produced by traffic-related and industrial activities. Strong epidemiological evidence supports a relationship between air pollution and the exacerbation of asthma and other respiratory diseases. Recent studies have suggested that air pollutants play a role in the development of asthma and allergies. Researchers have elucidated the mechanisms whereby these pollutants induce adverse effects; they appear to affect the balance between antioxidant pathways and airway inflammation. Gene polymorphisms involved in antioxidant pathways can modify responses to air pollution exposure. While the characterization and monitoring of pollutant components currently dictates pollution control policies, it will be necessary to identify susceptible subpopulations to target therapy/prevention of pollution-induced respiratory diseases.
INTRODUCTION
The global increase in the prevalence of allergic diseases is of great concern [1]. Among the numerous known causes of allergic diseases, urban air pollution has been attracting attention as an important environmental and extrinsic etiologic agent [2]. This includes gaseous materials such as ozone (O3) and nitrogen dioxide (NO2), as well as particulate matter (PM), which is generated by automobile traffic and industry. Strong epidemiological evidence supports a relationship between air pollution and the exacerbation of asthma and other allergic diseases; indeed, recent studies have suggested that pollutants play a crucial role in the development of asthma and other allergic disorders. Experimental studies have elucidated the cellular and molecular events that explain how these pollutants induce adverse effects in the respiratory system [3]. Additionally, recent gene-environmental approaches have been clarifying the mechanisms by which a host has increased susceptibility to air pollutants. This review will describe recent advances in our understanding of the impact of air pollutants on allergic diseases, with special reference to the adverse influence of particles [4].
AIR POLLUTANTS AND ALLERGIC DISORDERS: EPIDEMIOLOGICAL STUDIES
Increasing evidence supports a relationship between air pollutants and general respiratory symptoms and/or pulmonary function. Dockery et al. [5] found an association between air pollution and mortality in six US cities. Their prospective cohort study of 8,111 adults demonstrated that fine-particulate air pollution, or a more complex pollution mixture associated with fine particulate matter, contributed to excess mortality. This association was further strengthened by reports that reduced exposure to PM10 resulted in an attenuated age-related decline in lung function after 11 years [6,7]. Exposure to O3 at concentrations found in ambient air is associated with a reduction in lung function and induction of respiratory symptoms including cough and shortness of breath [8-11]. Importantly, individuals appear to differ with regard to susceptibility to O3 [11-13]. NO2 concentrations in ambient air are also reportedly associated with cough, wheezing, and shortness of breath in children. Gauderman et al. [14] found that urban air pollution may have lasting adverse effects on lung development in children aged 10-18 years, as measured by forced expiratory volume in one second (FEV1). Ackermann-Liebrich et al. [15] found that long-term exposure to air pollutants such as NO2 and PM10 in Switzerland was associated with diminished lung functioning in adults. Residential traffic-related air pollution exposure is reportedly associated with reduced expiratory flows in school children [16], and children relocating to areas with differing air pollution levels experience changes in lung function that mirrored changes in PM exposure [17]. Therefore, air pollution, especially pollution associated with traffic load, has a considerable impact on respiratory-related morbidity and mortality.
Many epidemiological studies have focused on the relationship between air pollution and allergic diseases, especially asthma.
Asthma exacerbation
Bronchial asthma is characterized by airway inflammation with dense infiltration of eosinophils and Th2 type lymphocytes, and increased hyperresponsiveness to non-specific stimuli as well as specific allergens [18]. Asthma is exacerbated by numerous causes including the common cold, excess allergen exposure, drugs, and changes in the weather [19]. Epidemiological studies have suggested a potential causal relationship between air pollution and exacerbation of asthma. NO2 exposure is associated with increased emergency room visits, wheezing, and medication use among children with asthma [20,21]. Controlled-exposure studies of asthmatics have found that NO2 can enhance the allergic response to inhaled allergens [22,23]. O3 exposure has also been linked to the frequency of hospital admissions [24,25], worsening of symptoms, and need for rescue medication [26] as well as asthma attacks, respiratory infections, and reductions in peak flow rate [27]. Many researchers have investigated the specific relationship between asthma and airborne traffic-related pollutants in urban areas. Traffic-related air pollution and respiratory symptoms are significantly associated in asthmatic children in Mexico City [28]: this study found that PM2.5, NO2, and O3 concentrations were significantly related to an increased incidence of exacerbated asthma. The authors also identified the impact of diesel-engine traffic on asthma wheezing and bronchodilator usage. Altered pulmonary function in children with asthma has been associated with highway traffic near their residence [29]. Among adults with asthma, exposure to traffic loads has been associated with lung function and health status [30].
This casual relationship between worsening asthma and diesel traffic pollution was clearly demonstrated by McCreanor et al. [31], who conducted a randomized, crossover study of 60 adult volunteers with either mild or moderate asthma. Each participant walked for two hours along a heavily trafficked street with high levels diesel (Oxford Street) and, on a separate occasion, walked for the same period through a nearby park with low levels of air pollution (Hyde Park). Walking along Oxford Street induced asymptomatic but significantly greater reductions in FEV1 (up to 6.1%) and forced vital capacity (up to 5.4%) than did walking through Hyde Park. This effect was greater in subjects with moderate asthma than in those with mild asthma. Importantly, these effects were accompanied by increases in biomarkers of neutrophilic inflammation (sputum myeloperoxidase) and airway acidification (maximum decrease in pH in exhaled breath condensates [EBC]). The changes were associated most consistently with exposure to ultrafine particles and elemental carbon. Therefore, exposure to air pollutants, especially particulate matter and certain gaseous agents such as NO2 and O3, exerts effects in the airways that ultimately result in asthma exacerbations [31,32].
Preliminary studies by our group [33] revealed a possible link between personal airway inflammatory biomarkers in EBC and air pollutant exposure. Measurement of EBC by R-tube is known to be a suitable method of assessing airway biomarkers because this method is safe and mobile and does not require electrical power [34]. A significant correlation appeared between the PM10 concentration one month before sampling and EBC and epidermal growth factor marker levels among asthmatics in Kawasaki, Japan. NO2 concentration was correlated with several markers in EBC in patients with asthma. EBC pH was significantly inversely related to the distance from main traffic-bearing roads. A sub-analysis was performed among moderate and severe asthma patients, and revealed that EBC biomarkers were correlated with PM10 concentration. This approach will facilitate the identification of individuals susceptible to air pollutants at the molecular level, which will in turn enable development of targeted therapy/prevention of pollution-induced respiratory diseases.
Impact of air pollutants on the development of asthma and allergy
In recent decades, the prevalence of allergic disease has dramatically increased globally, particularly in industrialized societies [35]. Genetic factors within populations, such as atopy, usually do not change in such a relatively short period, so researchers have postulated other causes for the increased prevalence of allergic diseases [36]. These include the so-called 'hygiene hypothesis', which refers to a lack of immunological shift from Th2-dominant, infantile states to Th1-dominant, adult type responses during the early period of life as a result of decreased exposure to bacterial infection [37]. Other researchers have focused on the role played by extrinsic agents, including air pollution [38]. Other than its effect on asthma exacerbation, the role of air pollution in the development of asthma in both children and adults remains unclear.
Nevertheless, recent studies, again with a focus on urban areas, have begun to report consistent associations. Three birth cohort studies from Germany, Holland, and Sweden, followed children until the age of four or six years and suggested a positive relationship between traffic-related pollution and physician-diagnosed asthma [39-41]. Two studies about to be published by the Californian Children's Health Study have found that traffic-related pollutants can cause asthma in older children. Gauderman et al. [14] reported that children exercising in a high O3 area were more likely to develop asthma, and Ackermann-Liebrich et al. [15] reported that a lifetime history of doctor-diagnosed asthma was associated with outdoor residential NO2 levels. Ryan et al. [42] found that exposure to traffic-related particles and endotoxin during infancy was associated with wheezing at age three among high-risk infants in Cincinnati. An Japanese cohort study also reported an association between NO2 levels and asthma incidence [43]; the authors studied the effects of air pollution on the prevalence and incidence of asthma among 2,506 children over a period of four years, and found that children living less than 50 m from heavily-trafficked roads were more likely to develop asthma. They also reported a possible link between asthma development and increased concentrations of PM10. Gehring et al. [44] found a positive association between traffic-related pollution levels at the birth address and incidence of asthma during the first eight years of life. Kunzli et al. [45] reported a correlation between traffic-related air pollution and adult-onset asthma among Swiss adults who had never smoked; they monitored 2,725 non-smokers aged 18-60 for 11 years (1991-2002) and found a significant relationship between asthma development and PM10 levels.
In contrast, Oftedal et al. [46] found no association between either modeled NO2 exposure or distance from a major road at the birth address and doctor-diagnosed asthma in a large cohort of Norwegian children. A more recent report from Phase One of the International Study of Asthma and Allergies in Childhood (ISAAC) [47] investigated the effect of ambient PM10 on the global childhood prevalence of asthma, rhinoconjunctivitis, and eczema using a World Bank model. Annual concentrations of PM10 at the city level were obtained for 105 ISAAC centers in 51 countries. After controlling for GNP per capita, a weak negative association was observed between PM10 and various outcomes. The findings suggest that urban background PM10 has little or no association with the prevalence of childhood asthma, rhinoconjunctivitis, or eczema either within or between countries.
Braback and Forsberg [48] summarized the results of nine large-scale prospective cohorts from 13 research projects. To date, all studies with the exception of the Norwegian report and the ISSAC study have reported a significant link between traffic-related pollution and the development of asthma.
Such apparent inconsistencies may have several explanations. Incomplete exposure assessment due to a lack of accurate data or variable lifestyles among residents might account for such results. For example, assessing exposure at the home address alone may neglect other activities such as school, where children spend most of the day and are physically active, which may increase their exposure to airborne pollutants. To address this issue, McConnell et al. [49] examined the joint effects of traffic-related pollution at home and at school and reported an association between the latter and an increased risk of asthma. Another important factor is gene-environmental interaction: an experimental study using a small number of patients reported that particular variants of oxidative stress genes, such as glutathione S-transferase (GST), play a crucial role in the effect of diesel exhaust particles (DEP) on allergic rhinitis [50]. Therefore, gene-related host susceptibility to air pollutants should be considered when a large scale cohort study is undertaken.
Susceptibility: Importance of gene-environmental interaction
It is important to identify subpopulations of asthmatics who are particularly susceptible to the effects of air pollution to ensure effective protection from adverse effects. Historically, host characteristics that influence the effects of air pollution on asthma include atopy, nutritional status, and social stress [51]. Traffic pollution affects asthma most strongly in children without a parental history of asthma [51]. This is consistent with reports that children without a family history of asthma were more likely to be diagnosed with asthma if they lived in a high-traffic area, compared to children with one or more asthmatic parents [52]. The observation that social stress and traffic-related air pollution are often spatially correlated has generated more research about possible synergistic effects on allergic disorders. Chronic stress has been found to modify the risk of asthma associated with traffic-related air pollution exposure [53]. Significant associations between higher lifetime NO2 exposure and asthma were found only among children who reported exposure to violence [54]. Stronger associations appeared between traffic-dense areas and respiratory symptoms among asthmatic children living in poverty [55] and between stress and inflammatory markers and respiratory symptoms among children exposed to lower NO2 levels [56].
Recently, researchers have focused on the relationship between air pollution and asthma and other allergic diseases from the perspective of gene-environmental interactions. The Cincinnati Childhood Allergy and Air Pollution Study birth cohort [57] was conducted to determine how environmental factors (DEP; environmental tobacco smoke, and mold) affect oxidative stress, which is thought to lead to persistent wheezing, and to determine how these effects are modified by the GST-P1 Ile105Val polymorphism. High levels of DEP exposure (> 0.5 µg/m3) were associated with an increased risk for wheezing only among the Val105 allele carriers. This finding demonstrates that gene-environmental correlations contribute to the effect of DEP on the development of persistent wheezing among infants with the GST-P1 Val105 allele. Additionally, nutritional supplementation with vitamins E and C influences the nasal inflammatory response to O3 exposure among asthmatic children [58]. This finding suggests that a diet rich in antioxidants may provide some protection against the adverse effects of ambient air pollutants. It may not, however, be feasible to prevent such effects with antioxidant supplementation during or just before a pollution event given the apparent complex control of antioxidant concentrations in the respiratory tract [59]. Romieu et al. [60] studied the GSTM1 polymorphism and how antioxidant supplementation affects lung function in relation to ozone exposure among asthmatic children in Mexico City. GSTM1-null children receiving a placebo showed significant ozone-related decrements in FEF (25-75), whereas GSTM1-positive children did not. Conversely, antioxidants had a stronger effect on children with the GSTM1-null genotype. The authors concluded that asthmatic GSTM1-deficient children may be more susceptible to the deleterious effects of ozone and might derive a greater benefit from antioxidant supplementation.
PARTICULATE MATTER AND ALLERGIES
Recent studies have investigated the adverse effects of particulate matter from heavy traffic load and industry. Special attention has been paid to the biological effects of DEPs, which are one of the main constituents of urban particulate air pollutants and are associated with allergic respiratory disorders, including asthma and allergic rhinitis [61]. Recent in vivo and in vitro studies strongly suggest that DEPs activate the transcription of both anti- and pro-inflammatory mediators.
Characteristics of DEPs
Diesel exhaust contains small particles that range in size from nanoparticles to coarse particles with an accumulation mass mode of 0.2 µm in diameter. Particles of this size have high deposition rates in the lung, and can persist in the atmosphere for lengthy periods [62]. These primary DEPs coalesce to form aggregates of a broad range of sizes and are important contributors to PM10 as well as PM2.5 in ambient air. DEPs consist of a carbonaceous core with a large surface area to which chemicals are adsorbed. These include organic compounds such as polycyclic aromatic hydrocarbons (PAHs), nitro derivatives of PAHs, oxygenated PAH derivatives (ketones, quinones and diones), heterocyclic compounds, aldehydes, and aliphatic hydrocarbons. PAHs and their oxygenated derivatives (e.g., quinones) have attracted special attention because they are able to redox cycle and generate reactive oxygen species (ROS) in target cells. The processes by which DEPs reach the cell surface and exert their influence have been partially elucidated [63-65].
Cellular and molecular mechanisms of the effects of DEPs
Ohtoshi et al. [66] reported that exposure to DEP in vitro stimulates human airway epithelial cells to produce cytokines relevant to airway inflammation. Bayram et al. [67] reported that exposure to DEP in vitro induced bronchial epithelial cells (BECs) to release interleukin-8 (IL-8), granulocyte-macrophage colony-stimulating factor (GM-CSF), regulated upon activation, normal T-cell expressed, and secreted (RANTES), and soluble intercellular adhesion molecules (ICAM)-1. BECs from asthmatic patients constitutively released significantly greater amounts of cytokines than did those from non-asthmatic individuals. DEPs up-regulated expression of the ICAM-1 gene in human BECs [68].
Research has also demonstrated that DEP-induced IL-8 production was regulated at the transcriptional level [69]. Additionally, DEPs have been observed to induce eotaxin gene expression in BECs [70], although another report refutes this [71]. DEPs induced dose-dependent activation of nuclear factor (NF)-κB in human BECs, as identified using a electrophoretic mobility shift assay [69]. Studies using reporter assays with normal and mutated IL-8 promoters indicated that IL-8 gene transcription was induced via NF-κB activation. Hashimoto et al. [72] reported that DEP-induced activation of p38 mitogen-activated protein kinase (MAPK) plays an important role in the production of IL-8 and RANTES. Other reports demonstrated the importance of other intracellular signal transduction pathways, such as mitogen-activated protein kinase kinase (MEK)-1 [73] and c-Jun N-terminal kinase (JNK) [74], in DEP-stimulated human BECs and macrophages.
Salvi et al. [75] studied the acute effects of high level diesel exhaust inhalation (300 µg/m3 for 1 hour) in a chamber in which healthy subjectss exercised continuously. They observed a significant increase in inflammatory cells (neutrophils, B lymphocytes, mast cells, CD4+, and CD8+ T lymphocytes) along with upregulation of ICAM-1 and vascular cell adhesion molecule-1. They also found that diesel exhaust inhalation led to increased levels of IL-8 and growth regulated oncogene-α, which are important for neutrophil accumulation [76]. They also attempted to clarify whether BECs from asthmatic individuals were more sensitive to DEPs than those from non-asthmatics with regard to production of pro-inflammatory mediators, but the data failed to reveal exaggerated airway inflammation in asthmatic individuals after diesel exhaust exposure [77,78]. Interestingly, epithelial staining for IL-10 increased after diesel exhaust exposure in the asthmatic group. These findings demonstrated that diesel exhaust exposure has evident inflammatory effects on the airways of non-asthmatic (or control) individuals, but that this does not occur in the presence of asthmatic airway inflammation. The role played by increased IL-10 levels after diesel exhaust exposure in the airways of asthmatic individuals warrants further investigation, because this cytokine can protect against inflammatory responses in other systems [79].
Other researchers have elucidated the molecular mechanisms underlying the diverse effects of DEPs in BECs. Zhang et al. [80] investigated the effects of DEPs on expression of fra-1, a heterodimeric partner of activator protein-1, in a murine lung epithelial cell line and found that DEPs markedly upregulated expression of fra-1 but not fra-2. Overexpression of fra-1 downregulated c-Jun, and nuclear factor-like 2 (Nrf-2) enhanced activator protein-1 and antioxidant response element mediated reporter gene expression, respectively. Fra-1 induction by DEPs may play a role in the selective regulation of expression of genes involved in alveolar epithelial cell injury and repair. Blanchet et al. [81] reported that PM2.5 and DEPs induced the expression and secretion of amphiregulin, an epidermal growth factor receptor (EGFR) ligand in BECs. Amphiregulin secretion was mediated by activation of the EGFR and extracellular signal-regulated kinase/MAPK pathways. Stimulation with amphiregulin induced GM-CSF release, and a neutralizing anti-EGFR antibody reduced particle-induced GM-CSF release. Exposure to diesel exhaust (PM10, 300 µg/m3) enhanced EGFR expression and phosphorylation of tyrosine 1173, which supports previous research about activation of the JNK, AP-1, p38 MAPK and NF-κB pathways and their associated downstream signaling and cytokine production [82]. These findings suggest that EGFR plays a key role in the bronchial response to diesel exhaust fumes. Long-term DEP exposure may lead to an airway remodeling process in asthmatics [83]. More profound adverse effects have also been reported, such as effects on the cardiovascular system [84].
Involvement of reactive oxygen species in the effects of DEPs
Notably, DEP-induced IL-8, ICAM-1, GM-CSF and RANTES expression is inhibited by antioxidant agents such as N-acetyl cysteine and pyrrolidine dithiocarbamate, so ROS may be involved in this induction [69,70]. Indeed, DEP-induced NF-κB activation was completely inhibited by pretreatment with NAC [69]. DEP-induced activation of MAPK pathways was also blocked by NAC and pyrrolidine dithiocarbamate [72]. These observations suggest that DEP-induced activation of signal pathways and transcription factors is a result of ROS, derived both primarily and secondarily from DEPs. Bonvallot et al. [85] found that the ability of DEPs to induce GM-CSF expression was almost completely eliminated after washing the DEPs, suggesting the importance of adsorbed chemicals. Benzene-extracted components had effects that mimicked those of DEPs on IL-8 gene expression, the release of several cytokines (IL-8, GM-CSF, RANTES), and NF-κB activation [86]. Tal et al. [87] reported that exposure to DEPs of varying organic content induces proinflammatory gene expression through multiple and differentially specific mechanisms in human airway epithelial cells.
In vivo animal models
A number of studies have shown that DEP exaggerates the airway inflammatory changes and airway hyperresponsiveness (AHR) induced by allergen sensitization followed by challenge [88]. Exposure of experimental animals to diesel exhaust enhanced AHR and allergic airway inflammation [89-91]. In conjunction with allergens, DEPs act as an adjuvant to enhance IgE antibody responses [92], inducing expression of cytokines/chemokines [93], and increasing AHR [94]. However, in studies of the potential of DEPs or diesel exhaust to induce toxicological effects, animals were generally exposed to a high dosage (i.e., ~3 mg/m3) [95], or via non-physiological routes (i.e., intratracheal injection of suspended particles).
Expanding on these findings, researchers investigated the effects of exposure to low-dose diesel exhaust (100 µg/m3 for 7 hours/day, 5 days per week, for 1-6 months) on AHR and allergic airway inflammation in asthmatic mice [96]. BALB/c mice were sensitized by intraperitoneal injection of ovalbumin and subsequently challenged by intranasal administration of ovalbumin. Repeated exposure of asthmatic mice to low-dose diesel exhaust resulted in increased AHR and expression of genes encoding several pro-asthmatic cytokines/chemokines.
Role of oxidative stresses induced by DEPs in vivo
Repeated low-level DEP exposure in two different strains of mice induced differential susceptibility to DEP exposure. This could be mediated by one or more of a number of anti-oxidant enzymes [97,98]. Nrf2 is a redoxsensitive basic leucine zipper transcription factor that is involved in the transcriptional regulation of many anti-oxidant genes [99]. Nrf2 is a key regulator of antioxidant defense in macrophages and epithelial cells and constitutes the main defense against the pro-inflammatory and oxidizing effects of DEPs (Fig. 1).
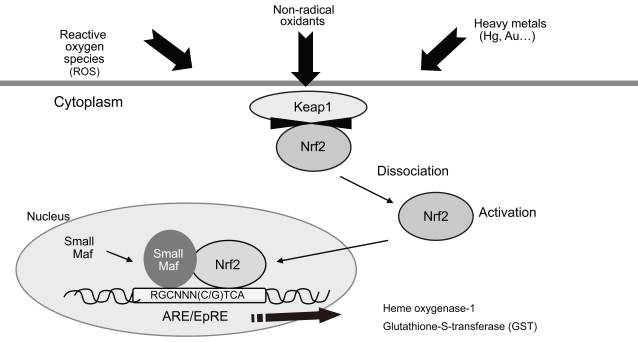
The transcription factor nuclear factor-like 2 (Nrf-2) plays an important role in cellular defense mechanisms against a variety of oxidant stresses, such as diesel exhaust fumes. ARE, antioxidant response element; EpRE, electrophile response element; Keap1, Kelch-like ECH-associated protein 1.
C57BL/6J Nrf2-/- mice exposed to low-dose DEPs for 8 weeks showed significantly increased AHR and lymphocyte and eosinophil counts, together with increased IL-12, IL-13, and thymus and activation regulated chemokine concentrations in bronchoalveolar lavage fluid than wild-type mice. In contrast, expression of antioxidant enzyme genes was significantly higher in wild-type mice than in Nrf2-/- mice [100]. These results strongly suggest that DEPs-induced oxidative stress and host antioxidant responses is regulated by Nrf2. The role played by Nrf2 in an allergic asthma model has been clearly demonstrated [100,101]; indeed, NAC treatment reduced the allergic inflammatory responses caused by DEP exposure [98]. Nrf2-/- mice exposed to low-dose DEPs exhibited significantly increased inflammatory cell numbers and periodic acid-Schiff staining-positive mucus cell hyperplasia. In contrast, expression of GSH/GSSG (reduced glutathione/oxidized glutathione) was greater in wild-type mice than in Nrf2-/- mice [102]. These results highlight the role of DEP-induced oxidative stress and host antioxidant responses in the exaggeration of allergic airway inflammation in mice. Furthermore, the responsiveness of the Nrf2-directed antioxidant pathway acts as a major determinant of susceptibility to allergen-mediated asthma [103]. These findings suggest that the synergistic effects of the oxidative stresses caused by DEPs and allergens contribute to the major pathways underlying exacerbation of allergic asthma.
Potential hazardous effects of Asian sand dusts
The potential exaggerating effects of so-called Asian sand dusts (ASD) on allergic diseases are of serious concern. These dusts originate from the deserts of Mongolia and China and spread to East China, Korea, Japan, and Taiwan, and occasionally even to North America. ASD have a diameter of 2.5 to 10 µm and contain a variety of bioactive substances, such as sulfates and nitrates. Some researchers have suggested that the increased PM10 level caused by ASD exposure is associated with exacerbation of asthma/allergic rhinitis [104]. In mouse models, ASD inhalation caused increased airway inflammation [105] and aggravation of allergic rhinitis [106]. The increased industrial development in China has sparked concern that ASD levels will increase and that its constituents will alter and possibly become more biologically active. The governments of China, Japan, and Korea are continuing to survey ASD contamination. Further studies will be necessary to elucidate the bioactivity of ASD in allergic diseases.
POSSIBLE CHEMOPREVENTION STRATEGY
Antioxidants reduce the allergic inflammatory effects of DEPs both in vitro and in a mouse model [107]. Romieu et al. [60] studied the influence of a GSTM1 polymorphism and antioxidant supplementation on lung function in relation to ozone exposure among asthmatic children in Mexico City. GSTM1-deficient asthmatic children appear to be more susceptible to the deleterious effects of ozone and therefore may benefit more from antioxidant supplementation. This finding strongly suggests that antioxidants may represent an effective prophylactic strategy against the adverse health effects of DEPs among susceptible populations. Tashakkor et al. [108] analyzed eight studies that investigated the role of antioxidant supplementation on measured lung function outcomes after exposure of subjects to air pollutants under controlled conditions. Five studies concluded that pollutant-induced airway hyperresponsiveness and diminution in lung function measurements were attenuated by antioxidant supplementation. Of 13 studies (from 12 publications), 10 reported attenuation of pollution-associated decrements. Therefore, increasing evidence supports the ability of anti-oxidant supplementation to moderate the effects of air pollution on lung function; however, more research using human subjects is necessary.
Based on these findings, it is important to develop methods of identifying susceptible individuals within a large population. Identification of GSTM1 polymorphisms is a possibility, but it would be costly and present ethical concerns. Future studies should evaluate whether measurement of airway inflammatory biomarkers in EBC would be an appropriate method of assessment.
CONCLUSIONS
Epidemiological, human, and animal studies all suggest that air pollutants (such as DEPs) are involved in the pathogenesis of allergic diseases such as asthma, both in terms of their development and exacerbation. Although these processes are still unclear, the adverse effect of air pollutants is unequivocal and avoidance measures should be implemented. Development of a safe and rapid method of screening individuals for their susceptibility to air pollution will allow effective chemoprophylaxis.
Acknowledgments
This work was supported in part by a grant from the Environmental Restoration and Conservation Agency of Japan.
Notes
No potential conflict of interest relevant to this article was reported.