Established and Newly Proposed Mechanisms of Chronic Cyclosporine Nephropathy
Article information
Abstract
Cyclosporine (CsA) has improved patient and graft survival rates following solid-organ transplantation and has shown significant clinical benefits in the management of autoimmune diseases. However, the clinical use of CsA is often limited by acute or chronic nephropathy, which remains a major problem. Acute nephropathy depends on the dosage of CsA and appears to be caused by a reduction in renal blood flow related to afferent arteriolar vasoconstriction. However, the mechanisms underlying chronic CsA nephropathy are not completely understood. Activation of the intrarenal renin-angiotensin system (RAS), increased release of endothelin-1, dysregulation of nitric oxide (NO) and NO synthase, up-regulation of transforming growth factor-beta1 (TGF-β1), inappropriate apoptosis, stimulation of inflammatory mediators, enhanced innate immunity, endoplasmic reticulum stress, and autophagy have all been implicated in the pathogenesis of chronic CsA nephropathy. Reducing the CsA dosage or using other renoprotective drugs (angiotensin II receptor antagonist, mycophenolate mofetil, and statins, etc.) may ameliorate chronic CsA-induced renal injury. This review discusses old and new concepts in CsA nephropathy and preventive strategies for this clinical dilemma.
INTRODUCTION
Cyclosporine (CsA) was first approved by the US Food and Drug Administration in the early 1980s for prophylactic anti-rejection therapy in patients receiving allogeneic transplants (kidney, liver, and heart). Its introduction has significantly improved both allograft and patient survival for over two decades [1,2].
The immunosuppressive action of CsA involves the intracellular interaction of CsA and calcineurin phosphatase, which reduces the production of interleukin-2 (IL-2). CsA initially binds to a specific family of receptors known as cyclophilins [3]. This drug-receptor complex inhibits the activation of calcineurin phosphatase, a secondary messenger in the dephosphorylation and activation of the nuclear factor of activation of T-cells (NF-AT). NF-AT is a regulatory protein that promotes the transcription and production of IL-2 and other cytokines that promote the growth and proliferation of T- and B-cells [4]. Inhibition of IL-2 production by CsA halts the proliferation and activation of helper and cytotoxic T-cells [5].
Despite the therapeutic benefits of CsA, several adverse effects have been reported in both transplant and non-transplant settings (i.e., autoimmune disorders), including toxicities (nephrotoxicity, hepatotoxicity, and neurotoxicity), hypertension, dyslipidemia, gingival hyperplasia, hypertrichosis, malignancies, and an increased risk of cardiovascular events [2,6]. The most clinically important complication is chronic CsA nephropathy, which is one of the known non-immunological factors causing chronic allograft nephropathy [7]. Chronic CsA nephropathy is characterized by progressive renal dysfunction, afferent arteriolopathy, inflammatory cell influx, striped tubulointerstitial fibrosis, and increased intrarenal immunogenicity [8-10]. Nankivell et al. [11] showed that almost all recipients presented evidence of chronic CsA nephropathy after 10 years of treatment with calcineurin inhibitors. The exact mechanism of this complication is not fully understood, although many potential mechanisms have been proposed.
Using a well-established animal model, we and other groups recently demonstrated that the mechanism of CsA-induced renal injury includes immunological and non-immunological pathways, as outlined in Figure 1. This review article summarizes our current understanding of the pathogenesis of chronic CsA nephropathy and discusses recent literature on the prevention and delay of this complication.
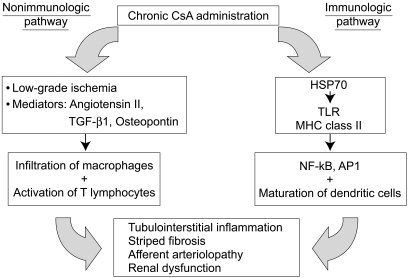
A schematic diagram of the pathogenesis of chronic CsA-induced renal injury. Chronic treatment with CsA induces renal injury by immunologic and non-immunologic pathways. The non-immunologic pathway includes hemodynamic (low-grade ischemia) and non-hemodynamic (mediators) mechanisms, although both ultimately result in tubulointerstitial inflammation, striped fibrosis, and an increased rate of chronic rejection. The immunologic pathway may be triggered by the endogenous production of HSP 70, a known activator of TLRs. Consequently, activation of the innate immune system by TLRs and MHC class II molecules leads to the activation of NF-κB and AP-1.
PREVIOUSLY ESTABLISHED MECHANISMS OF CsA-INDUCED RENAL INJURY
CsA-induced low-grade ischemia
Long-term administration of CsA rapidly decreases glomerular filtration rate (GFR) and renal blood flow by inducing vasoconstriction or increased intrarenal vascular resistance, which ultimately results in low-grade ischemic injury [2,6]. Chronic ischemia caused by CsA is believed to be associated with reactive oxygen species and lipid peroxidation [12,13]. This hypothesis is supported by the observation that antioxidant therapies significantly attenuate chronic CsA nephropathy [14,15].
The mechanism responsible for vasoconstriction is partially related to the activation of the intrarenal renin-angiotensin system (RAS) and imbalances in prostaglandins and thromboxane [13,16,17]. In addition, hypersecretion of endothelin-1 plays an important role in CsA-induced vasoconstriction [18]. Endothelin-1 is a potent vasoconstrictor, widely released in the kidney and vascular beds, and acts locally to increase vascular tone and regulate blood flow, GFR, and sodium reabsorption. Furthermore, endothelin-1 can also disrupt renal architecture through its effects on extracellular matrix (ECM) accumulation and tubulointerstitial fibrosis. However, after long-term withdrawal of CsA, both vasoconstriction and afferent arteriolopathy are shown to be normalize. CsA may also increase systemic vascular resistance associated with activation of the sympathetic nervous system [19].
CsA-induced activation of the renin-angiotensin system
Activation of the RAS, especially the intrarenal RAS, plays an essential role in the pathogenesis of chronic CsA nephropathy [20,21]. However, the mechanism of RAS activation in this complication remains unknown. One accepted hypothesis is that CsA increases renin release from juxtaglomerular cells. In a reproducible rat model of chronic CsA nephropathy developed by Rosen et al. [22] and Elzinga et al. [23], CsA administration to animals on a low-salt diet induced a significant decrease in GFR and histological changes similar to those described in patients undergoing long-term CsA therapy. Salt depletion activates the RAS, which is implicated in the changes in renal hemodynamics and function that follow CsA administration. Using this model, we and other groups clearly demonstrated that CsA significantly increases renin and angiotensin II (Ang II) immunoreactivity in the kidney [21,24,25], supporting a role for the intrarenal RAS in the pathogenesis of chronic CsA nephropathy.
Activation of the intrarenal RAS induces renal injury via hemodynamic and non-hemodynamic pathways. The RAS mediates vasoconstriction hemodynamically and thus leads to low-grade ischemia, as reviewed above. However, the RAS may also induce renal injury non-hemodynamically via stimulation of tubulointerstitial inflammation, the expression of transforming growth factor-beta 1 (TGF-β1) and vascular endothelial growth factor (VEGF), and increased renal cell apoptosis [26-29]. Blocking this system with either angiotensin-converting enzyme (ACE) inhibitors or angiotensin II (Ang II) receptor type I antagonists mitigates all of the above parameters and confers a renoprotective effect during chronic CsA nephropathy [21,25-28].
The pathogenesis of chronic CsA nephropathy may involve an association between the RAS and expression of the Klotho gene. The Klotho gene is an anti-aging gene involved in the suppression of several aging phenotypes [30]. Klotho gene expression in the kidneys is greatly reduced in patients with chronic renal failure [31] and is also suppressed in acute renal failure in ischemic-reperfusion injury murine models [32]. Recently, we found that the activation of the RAS suppresses Klotho gene expression in an animal model of CsA nephropathy and that blocking the RAS with Ang II receptor type 1 antagonists alleviates suppression of the Klotho gene and tissue injury (unpublished data). These results suggest that RAS activation suppresses Klotho gene expression and leads to chronic CsA-induced renal injury.
Nitric oxide and CsA-induced renal injury
In the kidney, nitric oxide (NO) is a vasodilating factor that plays a key role in maintaining vascular tone. In addition, NO decreases glomerular thrombosis and ischemia, mesangial cell proliferation, ECM protein synthesis, and interstitial inflammatory cell infiltration.
NO is produced from L-arginine by the action of NO synthase (NOS), of which at least three isoforms have been identified: neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS). These three NOS isoforms are all found in the kidney: nNOS is specifically expressed in macula densa cells, iNOS has been observed in mesangial and proximal tubular cells, and eNOS is expressed mainly in endothelial cells of the afferent and efferent arterioles and glomerular capillaries [33]. Chronic CsA treatment has been shown to differentially influence NOS isoform expression and interfere with NO production; however, contradictory results have also been reported [34-37].
The role of NO in the pathogenesis of chronic CsA nephropathy has been examined. We demonstrated that exogenous supplementation with L-arginine effectively prevents CsA-induced renal dysfunction, arteriolopathy, and interstitial fibrosis in rats [38]. Shihab et al. [39,40] also reported that L-arginine treatment attenuates CsA-induced TGF-β1 over-expression and ECM deposition, whereas these effects are reversed by treatment with N-nitro-L-arginine, a potent competitive inhibitor of NO biosynthesis.
Osteopontin as a pro-inflammatory cytokine in CsA-induced renal injury
The molecular mechanism underlying chronic CsA nephropathy is multifactorial. Because interstitial inflammatory events precede ongoing fibrosis [9], the up-regulation of chemoattractants and subsequent inflammatory cell infiltration are thought to play important roles in this condition.
Osteopontin (OPN) is a highly acidic phosphoprotein containing an arginine-glycine-aspartic acid (RGD) motif. It is involved in cell adhesion and migration [41] and is expressed by several cell types in a constitutive or inducible fashion. These include osteoclasts, some epithelial cells, macrophages, T-cells, smooth muscle cells, and tumor cells [42-47]. OPN acts as a chemotactic factor for macrophages and monocytes by binding to ligands such as αvβ3 integrin, CD44, collagen type I, and fibronectin [48,49]. The functional role of OPN, with respect to macrophage attraction, has been recently described in vivo and in vitro [41,50]. Moreover, the subcutaneous injection of OPN into rats produces massive macrophage accumulation, which is inhibited by the administration of an anti-OPN antibody [51].
In the kidney, OPN is expressed constitutively in the renal medulla in the loop of Henle and the distal convoluted tubules; it is absent in the normal renal cortex, with the exception of the parietal epithelium of Bowman's capsule. The up-regulation of OPN expression is strongly correlated with macrophage infiltration in several models of kidney diseases [28,52-55]. Young et al. [9] and Pichler et al. [28] reported that CsA treatment up-regulates OPN gene expression and is correlated with interstitial macrophage infiltration and fibrosis. We found that OPN mRNA and protein were constitutively present in the tubular epithelium, collecting ducts, and uroepithelial cells in control rat kidneys, whereas most cortical structures were negative for OPN. In contrast, the levels of OPN mRNA and protein increased dramatically in the tubular epithelium and Bowman's capsule cells in CsA-treated rat kidneys. The most striking change was observed in the renal cortex, which normally expresses very little constitutive OPN. Of note, the sites of strong OPN expression were in areas of macrophage influx and severe tubulointerstitial fibrosis [25,56]. Furthermore, a study in OPN-null mice demonstrated that the lack of OPN expression attenuated chronic CsA nephropathy [57]. These findings imply that OPN plays a pathogenic role in CsA-induced renal injury.
TGF-β1 as a pro-fibrotic cytokine in CsA-induced renal injury
TGF-β1 is a key cytokine implicated in the pathogenesis of a wide range of kidney diseases characterized by glomerulosclerosis and tubulointerstitial fibrosis, including chronic CsA nephropathy. Both in vivo and in vitro studies have shown that CsA administration is associated with dose-dependent increases in TGF-β1 expression. Shihab et al. [58,59] demonstrated that CsA-induced TGF-β1 up-regulation results in tubulointerstitial fibrosis, probably via its actions on ECM synthesis and degradation, and plasminogen activator inhibitor-1 plays a role in this process. Furthermore, administration of a specific TGF-β-neutralizing antibody ameliorated morphological alterations and preserved renal function in a mouse model of chronic CsA nephropathy [60].
TGF-β1 is secreted as a biologically inactive complex requiring in vivo activation. This latent TGF-β1 complex is activated via cleavage of its N-terminal latency-associated peptide to yield mature dimeric TGF-β1 through enzymatic and non-enzymatic mechanisms, or by the presence of the proteoglycan decorin and the scavenging protein α2-macroglobulin [61-65]. Therefore, increased amounts of TGF-β1 mRNA or protein may not actually represent parallel changes in its biologic activity.
Keratoepithelin (βig-h3) is a secreted matrix protein originally identified from a TGF-β1-stimulated human lung adenocarcinoma cell line (A549) [66]. βig-h3 has been proposed as one of the ECM components [67]; although the precise physiologic function of βig-h3 is unclear, it may connect various matrix components and resident cells, thereby serving as a bifunctional linker protein [68,69]. Thus, βig-h3 expression has been used to assess the biological activity of TGF-β1 [70]. Langham et al. [71] reported that βig-h3 production increased significantly in non-renal transplant recipients with chronic CsA nephropathy. More recently, we found that βig-h3 mRNA and protein were normally expressed in the cortex and outer medulla, specifically localized in the terminal portion of afferent arterioles (vascular pole of glomerulus), the S3 segment (parser recta) of the proximal tubules, and the distal convoluted tubules. However, in the CsA-treated rat kidney, βig-h3 gene expression was significantly up-regulated in the interstitium, but not in afferent arterioles or tubules, where interstitial expansion and fibrosis developed [72]. Thus, βig-h3 may be a useful index of TGF-β1 bioactivity and may reflect the degree of tubulointerstitial injury in chronic CsA nephropathy.
CsA-induced cell death
Tubulointerstitial injury is the prominent feature of chronic CsA nephropathy, and the major form of cell death is apoptosis [73]. Excessive loss of cellularity via apoptosis has been observed in fibrotic areas in renal biopsy specimens obtained from patients receiving long-term CsA therapy [74]. Apoptosis is an active mechanism of cell clearance and plays a key role in the regulation of cell number during development, in tissue homeostasis, and following insults. In the kidney, apoptosis may be beneficial [75] but is deleterious if enough resident cells are lost [76].
CsA has been shown to induce apoptotic cell death not only in T-lymphocytes, thus interfering with T-cell function [77], but also in some renal cells, resulting in the deterioration of kidney structure [26,78,79]. Thomas et al. [26] were the first to characterize the close link between apoptosis and interstitial fibrosis in a rat model of chronic CsA nephropathy. Subsequently, we and other groups have reported that CsA-induced renal cell apoptosis is associated with gene families such as the Bcl-2 proteins, Fas and Fas-ligand, p53, and the caspases [73,80], and that Ang II, NO, intrarenal growth factors (TGF-β1), epidermal growth factor (EGF), and macrophages are also involved [21,26,81-83].
CsA-induced activation of nuclear factor-kappa B and activating protein-1
Transcription factors such as nuclear factor-kappa B (NF-κB) and activating protein-1 (AP-1) regulate the gene expression of several cytokines, chemotactic proteins, adhesion molecules, and matrix proteins involved in inflammation, immunologic responses, cell differentiation, and the control of growth [84]. The transcription factors NF-κB and AP-1 are activated by a number of physiological and non-physiological stimuli such as cytokines, mitogens, viruses, mechanical factors, oxidative stress, and a variety of chemical agents [85-87]. Recent studies suggest that the activation of NF-κB and AP-1 is involved in the transcription of monocyte chemoattractant protein-1 and TGF-β1 in the kidney, which is regulated by Ang II or proteinuria [84,88-90]. In rats, Asai et al. [91] reported that the administration of CsA stimulated NF-κB and AP-1 DNA-binding activity, and this effect was blocked by the ACE inhibitor benazepril and by magnesium supplementation. In addition, we reported the involvement of the activation of NF-κB and AP-1 during the activation of innate immunity in chronic CsA nephropathy (described below) [92].
NEWLY DESCRIBED MECHANISMS OF CSA-INDUCED RENAL INJURY
Toll-like receptors and inflammation
The innate immune system primarily evolved as a rapid first line of defense, and is initiated by stimulation of toll-like receptors (TLRs) via their specific ligands [93,94]. Growing evidence indicates that the TLRs play a potential role in the pathophysiology of injury-associated kidney diseases [95]. Previously, we reported that CsA-induced renal injury produces endogenous TLR ligands such as heat shock protein (HSP) 70, up-regulates major histocompatibility complex (MHC) class II protein and TLR expression in renal tubular cells, and promotes the maturation of dendritic cells [92]. HSP 70 is an activator of TLRs and the innate immune system, and TLRs are recognized as sensors of pathogen-associated molecular patterns crucial for the initiation of an innate immune response [96,97]. Recently, we demonstrated that macrophage depletion decreased both TLR2 and MHC class II expression in renal tubular cells [83], which suggests a role for macrophages in immunologic injury. These results suggest the close association between the activation of innate immunity and chronic CsA-induced renal injury.
CsA-mediated impairment of urine concentration
CsA causes tubular dysfunction characterized by polyuria, calcium wasting, distal tubular acidosis, and hyperkalemia. Of these, impaired urine concentration is a predominant feature of chronic CsA nephrotoxicity [9], but the molecular mechanisms underlying this issue remain unknown.
In general, the urine-concentrating process is controlled by the aquaporin (AQP) system and the urea transporters (UTs) [98,99]. AQPs are a family of membrane proteins that play an important role in the reabsorption of water in the kidney. To date, at least 11 different types of AQPs have been cloned [98,100]. AQP1 aids in the rapid reabsorption of large quantities (70-80%) of filtered water and is constitutively present on both the apical and basolateral membranes of epithelial cells in the proximal tubule and the descending thin limbs of Henle's loop, and in endothelial cells of the descending vasa recta [101]. AQPs 2-4 are involved in the movement of water across the apical membrane in collecting duct principal cells and are localized to the basolateral membrane of this cell type. Reabsorbed free water determines the osmolality of the final voided urine, and this AQP-related process is controlled by vasopressin [98]. The UT family includes renal UT (UT-A) and the erythrocyte urea transporter (UT-B) [102]. In the kidney, UT-A1 and UT-A3 are responsible for the accumulation of urea in the inner medulla, and they are present in the inner medullary collecting duct. UT-A2 is present in the descending thin limbs of Henle's loop, and UT-B in the descending vasa recta [103-106]. We have demonstrated that chronic CsA treatment decreases AQP 1-4 and UT (UT-A2, UT-A3, and UT-B) production in rat kidneys, which may account for the impairment of urinary concentrating ability (polyuria, increased fractional excretion of sodium, and decreased free-water reabsorption) [107]. Recently, down-regulation of tonicity-responsive enhancer binding protein (TonEBP) has been proposed as a mediator of decreased AQP and UT transcription [108]. TonEBP is inhibited in the renal medulla secondary to reduced medullary tonicity, which is in turn the result of the down regulation of renal sodium transporters.
CsA-induced urinary calcium wasting
Another manifestation of renal tubular dysfunction caused by CsA is calcium wasting associated with the dysregulation of parathyroid hormone, which ultimately results in high bone turnover (osteopenia). Most of the filtered calcium (approximately 60%) is reabsorbed in the proximal tubule, and the remainder is reabsorbed in the medullary thick ascending limb of Henle's loop, the distal convoluted tubule, and the connecting segment [109]. The calcium binding protein calbindin plays an important role in calcium transport [110]. Two distinct subclasses of this protein with relative molecular masses of 9,000 and 28,000 Da have been recognized. Calbindin D9k is present in high concentrations in the proximal small intestine and facilitates intestinal calcium absorption [111]. Calbindin28k is expressed in the distal nephron segments of the rat kidney and is assumed to take part in calcium reabsorption [112]. Recent experimental studies by our laboratory [113] and by Steiner et al. [114] have demonstrated that chronic CsA treatment reduces calbindin28k immunoreactivity in the distal nephron, and this is accompanied by a significant decrease in serum calcium concentration and an increase in urinary calcium excretion. These results suggest that CsA-mediated suppression of calbindin28k production is a critical factor in renal calcium wasting.
Endoplasmic reticulum stress and autophagy
The endoplasmic reticulum (ER) has evolved a highly specific signaling pathway called the "unfolded protein response," a protective mechanism that reduces protein load and increases nascent protein folding, thus contributing to cell protection [115]. However, when ER stress is excessive and/or prolonged, the initiation of apoptosis is promoted [116,117]. Recently, Pallet et al. [118] demonstrated that CsA induces ER stress in renal tubular cells, which then activates autophagy as a protection against cell death [119]. Autophagy is a protective mechanism against various cellular stresses, including nutrient deprivation, hypoxia, and growth factor deprivation, and promotes cell survival. Recent evidence suggests that ER stress drives autophagy [120-122] and that autophagy alleviates ER stress and reduces cell death. We have demonstrated that prolonged ER stress induces apoptosis in an animal model of chronic CsA nephropathy [123]. Short-term treatment of CsA activated both the ER stress and proapoptotic responses, whereas long-term treatment of CsA decreased the ER stress response and increased the proapoptotic response. These results suggest that an imbalance between the two responses may cause apoptosis by depleting molecular chaperones and activating the proapoptotic pathway in chronic CsA-induced renal injury.
STRATEGIES TO PREVENT OR REDUCE CHRONIC CsA NEPHROPATHY
Reduction or withdrawal of CsA
Whether chronic CsA nephropathy is reversible is controversial. Traditionally, acute CsA nephropathy has been regarded as reversible after CsA dose reduction or complete withdrawal. However, chronic CsA-induced tubulointerstitial injury is considered to be persistent and, in some cases, progressive.
Weir et al. [124] investigated the long-term effects of the reduction or withdrawal of calcineurin inhibitors in a cohort of renal transplant recipients. After reduction or complete cessation of calcineurin inhibitors, a significant improvement in renal function was observed. Although an improvement in renal function was not recorded in all patients, this strategy was beneficial in most patients and reduced the rate of allograft loss in patients with nephrotoxicity. In low-salt diet-fed rats, Elzinga et al. [23] reported a progressive improvement in the GFR at 4 weeks after CsA discontinuation, but cortical fibrosis and tubular atrophy were unchanged. Using the same model, Franceschini et al. [125] carried out a long-term study in which the rats were administered CsA for 5 weeks, followed by the discontinuation of CsA for 2 or 8 weeks. The authors demonstrated that the degree of renal function and arteriolopathy was similar to that in placebo-treated controls after 8 weeks of CsA withdrawal, but tubulointerstitial fibrosis was not reversed. Therefore, there is dissociation between GFR and tissue injury in this animal model, and the low-salt diet is suggested to be an important factor.
To clarify the molecular mechanism of irreversible damage in chronic CsA nephropathy, we carried out two prolonged studies using two dosages of CsA (7.5 and 15 mg/kg) and multiple periods of drug washout [126]. Surprisingly, both renal function and morphological alterations (tubulointerstitial fibrosis and arteriolopathy) were reversed in rats with both dosages of CsA at 5 weeks after withdrawal, and the alleviation of tubulointerstitial fibrosis was observed after 10 weeks. At the molecular level, the reversibility of chronic CsA nephrotoxicity was closely associated with the down-regulation of OPN and TGF-β1 gene expression, the up-regulation of EGF production, and a decrease in renal cell apoptosis [82]. These conflicting results between our studies and previous reports lead us to speculate that the effect of CsA withdrawal in chronic CsA-induced renal injury may depend on the CsA dosage or on the timing of drug elimination.
Given the recently demonstrated association between prolonged ER stress and apoptosis [123], kidney allografts may well adapt to CsA-induced ER stress by inducing the expression of molecular chaperones. The duration and intensity of CsA-induced ER stress may provide a rationale for the reduction or withdrawal of CsA to prevent chronic CsA nephropathy. In addition, because the activation of autophagy indicates that cellular stress has occurred, the early detection of autophagy may facilitate the timely reduction or withdrawal of CsA treatment [119]. Clinical trials are needed to clarify these issues.
Pharmacologic intervention
Chronic CsA nephropathy is a major problem in transplant recipients and patients with autoimmune disorders, as it may lead to end-stage renal disease requiring dialysis. Therefore, renal function should always be carefully monitored, even in patients in whom renal function appears to be stable. In addition, early recognition and pharmacological intervention may be necessary to minimize the rate of allograft loss or chronic renal failure.
It is generally accepted that the modulation of vasoactive factors (Ang II and NO) effectively prevents CsA-induced renal injury. Previous studies clearly demonstrated that the concomitant administration of ACE inhibitors and Ang II type I receptor antagonists significantly reduced arteriolopathy, interstitial fibrosis, and tubular atrophy, independently of hemodynamic effects. However, renal dysfunction was not attenuated, and a potential explanation for this discrepancy has been discussed above. The molecular mechanisms underlying the renoprotective effects of these drugs in chronic CsA nephropathy may be related to their actions on inflammatory mediators, pro-fibrotic cytokines, matrix proteins, apoptotic cell death, and innate immunity [9,16,21,26-28,127). BQ123, an endothelin receptor blocker, partially improved renal function, but did not reduce structural damage [128]. Similarly, exogenous supplementation of L-arginine, a substrate for NO synthase, also conferred a renoprotective effect [38,40]. This effect was associated with the reduction of VEGF expression and the increased availability of NO.
Drugs with anti-fibrotic properties such as the TGF-β antibody [60], statins [129], pirfenidone [59,130], and hepatocyte growth factor [131] have been shown to have renoprotective effects in chronic CsA nephropathy. Statins are competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the key enzyme that regulates the synthesis of cholesterol from mevalonic acid by suppressing the conversion of HMG-CoA [132]. The inhibition of HMG-CoA reductase by statins has pleiotropic effects independent of their lipid-lowering effects (e.g., anti-inflammatory and anti-arteriosclerotic effects) [133,134]. These benefits are mirrored in many renal disease models [135-138]. In a rat model of chronic CsA nephropathy, we showed that pravastatin treatment attenuated interstitial inflammation and fibrosis. The suppression of OPN, TGF-β1, and intrarenal C-reactive protein expression along with increased eNOS expression may be responsible for the renoprotective properties of pravastatin [129].
Anti-inflammatory drugs, such as pentosan polysulfate [139] and mycophenolate mofetil (MMF) [25], are also suggested to have protective effects in chronic CsA-induced renal injury. In a study by Schwedler et al. [139], pentosan polysulfate reduced tissue injury but was unable to improve renal function. We have reported that the administration of MMF is partially effective in preventing the development of chronic CsA nephropathy. Moreover, the combination of MMF and losartan (an Ang II type 1 receptor antagonist) or the administration of MMF after CsA withdrawal affords superior protection compared to losartan monotherapy [25] or CsA withdrawal alone [140].
Other strategies include vitamin E [14], recombinant human erythropoietin (rHuEPO) [141], rosiglitazone [142,143], or spironolactone [144,145] therapy. Vitamin E treatment is reported to preserve renal function and reduce free radical production, vasoconstrictive thromboxane levels, and tubulointerstitial fibrosis, probably by suppressing the production of superoxide anion, hydrogen peroxide, malonyldialdehyde, hemeoxygenase I, and some mediators (TGF-β1 and OPN) [14]. The preventive effect of rHuEPO on chronic CsA-induced renal injury is associated with its anti-apoptotic and anti-inflammatory properties [141]. The exogenous administration of rosiglitazone, a peroxisome proliferatoractivated receptor gamma agonist, has been shown to reduce chronic CsA-induced renal injury [142,143], but the mechanism underlying this effect remains unknown. The blockade of aldosterone receptors with spironolactone has been shown to ameliorate chronic CsA nephropathy [144,145]. Interestingly, spironolactone therapy not only attenuated structural damage, but also improved renal function, perhaps via the prevention of TGF-β and extracellular matrix protein over-expression and the reestablishment of renal blood flow.
CONCLUSION
To reduce or avoid chronic CsA-induced renal injury, new immunosuppressive agents and strategies for calcineurin inhibitor minimization, avoidance, or withdrawal have been emerging in the literature. These new strategies may ultimately minimize the incidence of nephrotoxicity and improve allograft and patient survival. Clinical trials are warranted to investigate the appropriate dosage of CsA and the effect of eliminating CsA with the addition of non-nephrotoxic immunosuppressants, such as MMF and sirolimus, which may permit optimal immunosuppression while avoiding the risk of acute rejection. However, CsA is still a major immunosuppressant currently used in both transplant and non-transplant patients. Considering the increasing rate of late allograft loss or chronic renal failure associated with chronic CsA nephropathy, a full understanding of the molecular mechanisms underlying chronic CsA-induced renal injury is essential to improve long-term outcomes and pharmacological interventions to delay the progression of chronic CsA nephropathy.