Pathogenetic Role of Thyrotropin Receptor Antibody in the Development of Hyperthyroidism Following Primary Hypothyroidism*
Article information
Abstract
The authors measured thyrotropin binding inhibitory immunoglobulin (TBII), thyroid stimulating antibody (TSAb), and thyroid stimulation blocking antibody (TSBAb) sequentially in patients who developed hyperthyroidism following primary hypothyroidism, and compared changes in these various funcional parameters of thyrotropin receptor antibody (TRAb) with clinical manifestations, in order to investigate the role of TRAb in the development of hyperthyroidism following primary hypothyroidism.
In a patient with goitrous chronic thyroiditis, TBII, TSAb and TSBAb were not detected at the initial hypothyroid phase. But with appearance of TBII and TSAb, the patient developed hyperthyroidism. In a patient with primary nongoitrous myxedema, initially high TBII and TSBAb were detected without TSAb activity. His TSBAb disappeared and TSAb appeared with development of goiter growth and hyperthyroidism.
These two mechanisms, that is, appearance of previously absent TSAb and conversion of TSBAb to TSAb, might play a causative role in the development of hyperthyroidism following primary hypothyroidism. These phenomena might be evidence that Graves’ disease, chronic thyroiditis, and primary nongoitrous myxedema are on a continuing spectrum of a common syndrome sharing similar pathophysiology, at least with respect to TRAb.
INTRODUCTION
Chronic autoimmune thyroiditis usually runs a stable course, and only occasionally do profound changes in functional status occur.1,2) There are, however, several well documented cases of hyperthyroidism which developed spontaneously from primary hypothyroidism.3,4,5) About 40 cases are reported in the English literature5), but it is uncertain how often this unusual phenomenon occurs and what is the exact pathogenetic mechanism. Obviously, autoimmunity plays a major role6), and thyrotropin receptor antibody (TRAb) might play a particularly important role. That is, previously nonexistent thyroid stimulating antibody (TSAb) develops in a patient with chronic thyroiditis and stimulates remaining follicular epithelial cells to proliferate and hyperfunction, resulting in hyperthyroidism.7) Alternatively, in thyroid stimulation blocking antibody (TSBAb) associated primary nongoitrous myxedema, TSBAb somehow changes to TSAb, resulting in sustained stimulation of the follicular cells causing hyperthyroidism.8)
There is no doubt that TSAb causes hyperthyroidism in Graves’ disease.9,10) TRAb is generally not pure TSAb, but is a compound mixture of heterogeneous antibodies, differing in biological characteristics. In Graves’ disease, TSAb disappears and TSBAb appears with development of hypothyroidism after radioiodine therapy11,12) or even after antithyroid drug treatment.13,14,15) Moreover, once developed hypothyroidism with emergence of TSBAb reconverts to Graves’ hyperthyroidism with disappearance of TSBAb and reappearance of TSAb.16,17) The above findings suggest that the biological character of TRAb determines the clinical manifestations in autoimmune thyroid diseases.
In this study, we serially measured thyrotropin binding inhibitory immunoglobulin (TBII), TSAb, and TSBAb when hyperthyroidism developed following primary hypothyroidism, and compared the various functional parameters of TRAb with clinical status, to clarify the role of TRAb in this unusual phenomenon.
MATERIALS AND METHODS
1. Subjects
Chronic thyroiditis was diagnosed when a patient presented with diffuse goiter, elevated serum TSH level, and positive thyroid autoantibodies. Primary nongoitrous myxedema was diagnosed when another patient presented with clinical hypothyroidism, impalpable thyroid, low serum T4, elevated serum TSH, and decreased 24h radioactive iodine uptake. Hyperthyroid Graves’ disease was diagnosed clinically based on the findings of clinical symptoms, diffuse goiter, elevated serum T3 and T4, decreased TSH, and increased thyroidal radioactive iodine uptake, which was not suppressed by T3 administration.
Serum samples were stored in aliquot at −70°C until use. IgG was prepared by means of affinity chromatography using protein A-Sepharose CL-B (Pharmacia, Sweden).
2. Thyroid Function Test and Assay for Thyroid Autoantibodies
Twenty-four hour thyroidal radioiodine uptake was measured by the standardized method. Serum T3BU, total T3, and total T4 were measured by commercially available RIA kits from Abbott (USA). Serum TSH was measured by ultrasensitive immunoradiometric assay using kits from Abbott (USA), and the normal range was 0.4–4.1 μu/ml.
Antimicrosomal antibody and antithyroglobulin antibody were measured by radioimmunoassay using kits from R.S.R. Ltd (UK) and values above 3U/ml were regarded as positive.
3. Assay for TBII
TBII was measured as described previously18) using commercial radioreceptor assay kits from R.S.R. Ltd (UK). TBII activity was expressed as percent inhibition of radiolabelled bTSH binding to its receptor and values above +15% were regarded as positive.18)
4. Assay for TSAb and TSBAb
FRTL5 cells, generously donated by Dr. Kohn at NIH, USA, were maintained as previously described.19) After 7 days without TSH, 300μl of IgG (10mg/ml) was added to each well and incubated at 37°C, in 5% CO2-95% air, for 2 hours. The cAMP released into culture supernatant was measured by RIA (Immunonuclear, Still Water, MN, USA). TSAb activity was expressed as percent increase in cAMP production by test IgG compared to normal control IgG. Values above 170% were considered positive.19)
When measuring TSBAb, IgG was incubated with or without 0.1 mU/ml bTSH. Other procedures were the same as the TSAb assay. TSBAb activity was expressed as percent inhibition of 0.1 mU/ml bTSH induced cAMP production by test IgG compared to normal control IgG. Values above 37% were considered abnormal.20) In these bioassay systems, intra-assay variance was 5.0–7.4% and interassay variance was 17.0–32.5%.19)
RESULTS
1. Patient 1
A 29-year-old female visited the Thyroid Clinic at Seoul National University Hospital with the chief complaint of a recently growing diffuse goiter in March 1985. No subjective symptoms were found. On examination, a diffuse goiter of moderate size (about 60gm) was the only abnormal finding. Laboratory examination showed T3RU 27.4%; T3 126ng/dl; T4 6.9μg/dl; TSH 5.8μu/ml; antimicrosomal antibody 80U/ml; and antithyroglobulin antibody below 3U/ml. TBII, TSAb and TSBAb were all negative with values of 5.8%, 110%, and 5.1% respectively. She received 100μg of ℓ-thyroxine daily, with a decrease in goiter size (about 30gm) after two months. The thyroxine dose was increased to 150μg per day and continued.
In December 1985, TSAb activity appeared but she was euthyroid. In March 1986, the goiter size increased (about 50gm) with development of perspiration, palpitation and hand tremor, and a thyroid function study revealed T3RU 46.2%; T3 360ng/dl; T4 22.0μg/dl; and TSH below 0.05μU/ml. Antimicrosomal antibody was 20U/ml, and antithyroglobulin antibody was negative.
The 24 hour thyroidal radioiodine uptake was 56.5% and was not suppressed by T3 administration. At this time, TBII was 23.7 %, and TSAb 611 %, but TSBAb was persistently negative. Thyroid needle biopsy showed diffuse epithelial hyperplasia compatible with Graves’ disease (Fig. 1). Antithyroid medications were started and thyroid function normalized with a decrease in TBII and TSAb. TBII and TSAb disappeared ultimately and she remains euthyroid clinically and biochemically after withdrawal of the antithyroid drug until March 1988 (Fig. 2).
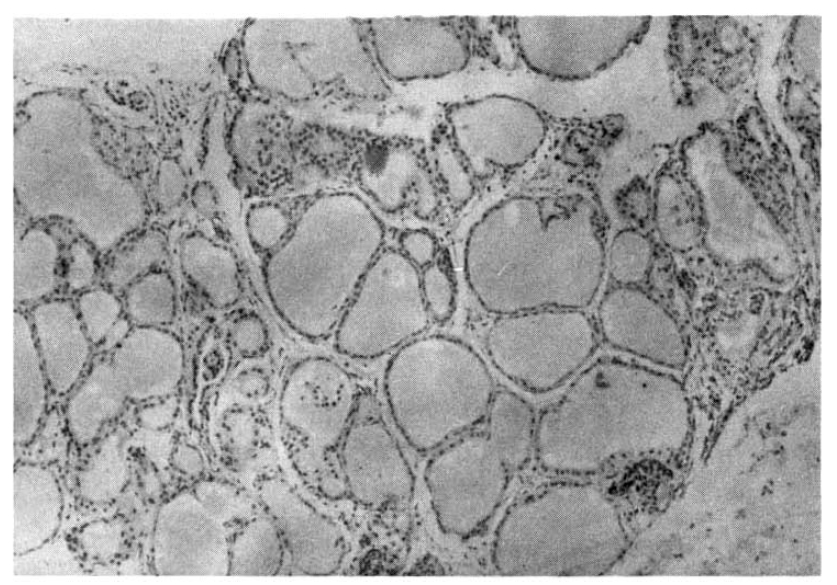
Thyroid needle biopsy performed when patient 1 became hyperthyroid shows diffuse epithelial hyperplasia compatible with Graves’ disease.

Clinical course and changes in TRAb in patient 1. Upper panel (○) TSH, (•) T4; lower panel (○) TSAb. (•) TBII. PTU: propylthiouracil.
2. Patient 2
A 40-year-old male visited the Thyroid Clinic at Seoul National University Hospital due to fatigue, myalgia, and diplopia in November 1986. The thyroid was impalpable. Except for bilateral ptosis and a slightly puffy face, there was no abnormal physical finding. Technetium thyroid scan revealed no demonstrable thyroid uptake, and 24 hour thyroidal radioiodine uptake was 2.1%. Serum T3BU was 28.5%, T3 90ng/dl, T4 4.2μg/ml, and TSH 122μU/ml. Antimicrosomal antibody was 10U/ml, and antithyroglobulin antibody was negative.
TBII was 80.2%, TSAb was negative with 91.9% but TSBAb was very high with a value of 99.4%. Tensilon test was positive. He was regarded to have primary nongoitrous myxedema probably due to TSBAb and myasthenia gravis and was treated with 100μg of ℓ-thyroxine and 300mg of pyridostigmine daily. His clinical symptoms soon disappeared and thyroxine was increased up to 150μg per day. In March 1987, myasthenic symptoms were aggravated despite increased pyridostigmine, and prednisolone was tried since April 1987. At that time, his thyroid function was normal with 150μg thyroxine, and TRAb activities were the same as those at diagnosis.
During the slow tapering of prednisolone, he lost about 5kg in weight, and palpitation, sweating, and hand tremor appeared in December 1987. Thyroxine was stopped immediately but symptoms persisted, and a previously undetected diffuse goiter was noted (about 40gm). In January 1988, a thyroid function study revealed T3BU 40.0%; T3 395ng/dl; T4 19.0μg/dl; and TSH below 0.05μU/ml. His 24 hour radioiodine uptake was 56.6% and was not suppressed by T3. TBII was 45.1 %. TSAb was 2703%, an exceedingly high activity, and the previously positive TSBAb was 24.9%, within normal range. Eye symptoms were not prominent and no exophthalmos was noted. He was treated with antithyroid drugs. Thyroid function became normalized with further decreases in TBII and TSAb. TSBAb did not appear and goiter size did not change (Fig. 3).
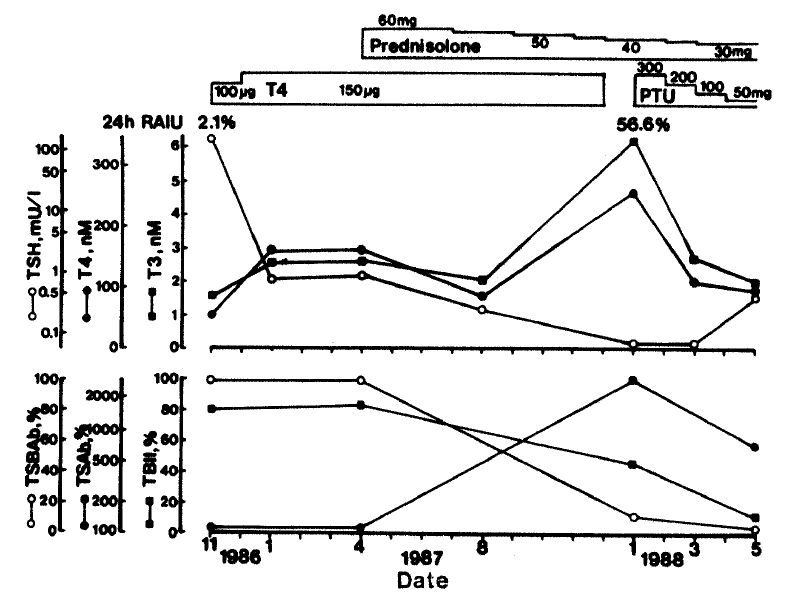
Clinical course and changes in TRAb in patient 2. Shaded areas represent normal ranges. Upper panel (○) TSH, (•) T4, (▪) T3. Lower panel (○) TSBAb, (•) TSAb, (▪) TBII. PTU: propylthiouracil.
DISCUSSION
Graves’ disease, primary nongoitrous myxedema, and chronic lymphocytic thyroiditis are thought to be on a continuing spectrum of a syndrome sharing a common etiopathogenesis.10) Clinical Graves’ hyperthyroidism with histological Hashimoto’s thyroiditis is known,21) and sometimes Graves’ disease evolves spontaneously into hypothyroidism due to concomitant chronic thyroiditis.13,22) Hypothyroidism following Graves’ hyperthyroidism is observed frequently1) but the reverse is very rare and the pathogenic mechanism is more obscure.
One possible mechanism is “Jod-Basedow syndrome” which is a phenomenon of hyperthyroidism fueled by iodine replacement in hypothyroid patient living in iodine-deficient areas.23) In our patients, iodine intake or urinary iodine excretion was not measured. Nowadays, however, there are no iodine-deficient areas in Korea and furthermore, in these patients, there was no history of excessive intake of iodine-containing drugs or foods. It seems unlikely that Jod-Basedow syndrome played a significant role in the development of hyperthyroidism in these patients.
Reviewing the literature, TRAb activity was detected at the hyperthyroid phase, although sequential measurements were not performed in most of the cases, (3,4,5,6,7,8,16,24,25,26,27) and functionally stimulating antibody was detected. The demonstration of TSAb at the hyperthyroid phase seems quite appropriate, in that, more than 90% of patients with fresh untreated Graves’ disease have detectable TSAb.9,10) But clear documentation of the whole sequence is rarely reported,7) and in some cases, TSAb activity was present at the initial hypothyroid phase.28) The presence of TSAb is associated with the development of hyperthyroidism with certainty, but the degree of tissue destruction might determine the chance of hyperthyroidism developing. If relatively enough follicular epithelial cells are preserved, TSAb might stimulate these cells, resulting in hyperthyroidism. In contrast, a severely destroyed thyroid cannot be stimulated even in the presence of TSAb.
In our patient 1, it was clearly documented that previously absent TSAb appeared, then hyperthyroidism developed. Moreover, the appearance of TSAb preceded development of hyperthyroidism. These findings are entirely compatible with the above statements that emergence of TSAb stimulates the remaining cells, resulting in hyperthyroidism.
At present it is unclear why TSAb develops during the course. Since thyroid hormone itself may directly affect suppressor T lymphocyte function,3) and in most of the patients reported, thyroid hormone therapy was done before the development of hyperthyroidism, thyroid hormone treatment might be regarded to trigger hyperthyroidism. But of the enormous number of patients receiving thyroid hormone, only the occasional patient develops hyperthyroidism. In our patient 1, the administered dose was within the physiologic range. Moreover, there are occasional cases who developed hyperthyroidism without previous thyroid hormone therapy.5,29,30) It is unlikely, therefore, that thyroid hormone causes or contributes to this phenomenon.
In the course of subacute thyroiditis, TRAb is detected in most of the patients, at least transiently, and this phenomenon is regarded as secondary immune response to tissue destruction and antigenic release.31) Likewise, in the course of chronic thyroiditis, transient aggravation of †issue destruction with antigenic release might form TSAb producing clones. It can be assumed that in some genetically predisposed patients, immune dysregulation fails to abolish TSAb producing clones resulting in Graves’ hyperthyroidism, but still no evidence exists.
TSBAb is frequently found in patients with primary hypothyroidism, especially in primary nongoitrous myxedema, and is regarded to play a pathogenetic role in primary nongoitrous myxedema.20) TSBAb activity in primary myxedema usually does not change with time or therapy,20) in contrast with TSAb in Graves’ disease which decreases after antithyroid drugs10,19,32) or surgery.10,33) Only rarely does TSBAb disappear with spontaneous recovery of thyroid function,34) and disappearance of TSBAb associated with emergence of TSAb and hyperthyroidism is reported extremely infrequently.8,16,17)
In our patient 2, the initial TSBAb activity was so high that it almost completely inhibited the action of 0.1 mU/ml bTSH. Such potent TSBAb activity disappeared somehow during the course and TSAb appeared with hyperthyroidism and goiter growth. He also had myasthenia gravis and steroid treatment was administered. Since this event occurred during prolonged high dose steroid therapy, there might be an association. But the exact cause is uncertain at present. It may be considered that TSAb producing clones were present in addition to TSBAb producing clones at the initial hypothyroid phase and that steroid treatment selectively removed TSBAb producing clones, since steroid treatment decreased TSBAb activity in a patient with primary myxedema.35) Considering the profound nonspecific immunosuppressive effects of high dose steroid, however, it is unlikely that only TSBAb producing clones were selectively removed. In this patient, two receptor antibody mediated diseases coexisted; that is, acetylcholine receptor antibody mediated myasthenia gravis and thyrotropin receptor antibody mediated primary myxedema, suggesting more severe immune dysregulation. Perhaps TSAb producing clone appeared spontaneously after removal of TSBAb producing clone due to preexisting severe immune dysfunction.
In this study, we ascertained that clinical thyroid function status can be changed according to the character of TRAb.36,37) TRAb plays an important central role in the development of hyperthyroidism following primary hypothyroidism. Previously absent TSAb appears with resultant hyperthyroidism, or TSBAb somehow changes to TSAb, resulting in hyperthyroidism. At least these two mechanisms might be associated with development of hyperthyroidism following primary hypothyroidism, and these phenomena are considered to be evidence that Graves’ disease, chronic thyroiditis, and primary nongoitrous myxedema are on the spectrum of a syndrome sharing a common pathogenetic mechanism.
Acknowledgements
We are grateful to Miss JH Choi who helped us to prepare manuscript.
Notes
This study was supported by a Clinical Research Grant from Seoul National University Hospital