Rapidly Progressing Budd-Chiari Syndrome Complicated by Hepatocellular Carcinoma
Article information
Abstract
Budd-Chiari syndrome (BCS) is a disorder caused by occlusion of the hepatic vein or inferior vena cava. The clinical presentation include abdominal pain, hepatomegaly, ascites, leg edema, collateral venous dilatation of the body trunk, and portal hypertension. In addition, BCS can cause hepatocellular carcinoma (HCC) in some patients, although its pathogenesis is not yet completely understood. The average reported time lag from diagnosis of BCS to full-blown HCC ranges from several years to several decades. Hepatic carcinogenesis in patients with BCS perhaps reflects a prolonged and persistent liver injury in that it occurs in the primary inferior vena cava obstruction rather than the primary hepatic vein thrombosis. Among patients with BCS, membranous obstruction of the vena cava (MOVC) usually presents an insidious and chronic illness, whereas primary hepatic vein thrombosis presents an acute or subacute illness. We experienced a case pf a patient with BCS, which progressed rapidly that HCC developed only nine months after the diagnosis of BCS. The factors causing this rapid progression are still unclear and remain to be investigated.
INTRODUCTION
HCC is one of the more devastating malignant diseases in the world. Its etiology and frequency in a population varies markedly in different geographic areas, which means that chronic viral infections such as hepatitis B and C play a major role in its development. Cirrhosis of the liver, regardless of its etiology, has been recognized as a predisposing condition for several decades, and factors such as hepatitis B and C viruses, alcohol consumption, hemochromatosis, tyrosinemia, exposure to oral contraceptives, anabolic steroids, and aflatoxins determine, in part, the prevalence of HCC. BCS is also known as a minor predisposing factor in the development of HCC1, 2).
Although there are many reports stating that BCS occurs as a complication of HCC, cases of plain HCC occurring as a complication of BCS have been reported less frequently3). As a result, there are few reports that characterize and analyze HCC arising from BCS. Recently, we experienced a 37-year-old female patient with BCS complicated by HCC that arose only nine months after the diagnosis of BCS. To our knowledge, this patient’s progression to HCC was more rapid than any other cases previously reported. We describe here the characteristics of this case and consider the possible factors related to this rapid progression from BCS.
CASE REPORT
A 37-year-old woman was admitted to Kangnam Saint Mary’s Hospital with about month-long history of abdominal discomfort and leg edema. She had no previous record of serious diseases. She avoided excessive alcohol consumption and smoking, but she had been drinking five to six cups of Chinese medical tea per day for weight reduction and chronic constipation. Her height was 160 cm and her weight was 64 kg. On physical examination, her abdomen was mildly distended and pitting edema was noted on both lower legs. Initial laboratory blood tests showed a white blood cell count of 3,800/mm3 with 40% neutrophils, hemoglobin concentration 11.4 g/dL, a platelet count of 107,000/mm3, serum aspartate aminotransferase to alanine aminotransferase (AST/ALT) 42/58 IU/L, total bilirubin concentration 1.72 mg/dL, total protein 6.8 g/dL, albumin concentration 3.4 g/dL, alkaline phosphatase level 158 IU/L, γ-GTP concentration 47 IU/L, prothrombin time 89%, and α-fetoprotein concentration 2.7 ng/mL. All the other data were within normal values. Serum markers for autoimmune, hepatitis B and C, and human immunodeficiency virus (HIV) were all negative. Coagulation tests including protein C, protein S, and anti-thrombin III were also within normal ranges. The patient underwent abdominal dynamic computerized tomography (CT) scan (Figure 1A) and magnetic resonance imaging (MRI) (Figure 1B), which revealed a hypertrophied caudate lobe of the liver, splenomegaly, obliterated hepatic veins, and the narrowing of the intrahepatic vena cava. Venography (Figure 2) showed a broad stenosis of the intrahepatic vena cava consistent with the CT and MRI findings. There was no radiological evidence of hepatic mass. Liver histopathology obtained by a laparoscopic biopsy revealed zone 3 hemorrhage due to hepatic congestion, chronic inflammatory cell infiltration in the portal area, and fibrotic nodular formation (Figure 3). Two months later, her ascites became uncontrollable despite vigorous diuretic treatment, necessitating mesocaval shunt surgery. Under the surgical operation, there was no evidence of hepatic mass or intra-abdominal lymphadenopathy. Three weeks later, she was discharged with an improvement in her condition and followed up at an out-patient clinic. Seven months after she was discharged, she was admitted again with complaints of general weakness, nausea, and abdominal discomfort. A physical examination showed a slightly icteric sclerae, a mildly distended abdomen, and splenomegaly to a breadth of three fingers. Blood tests showed a total bilirubin concentration of 2.9 mg/dL, albumin 2.6 g/dL, AST/ALT 88/96 IU/L, prothrombin time 70%, and α-fetoprotein 168.4 ng/mL. The patient was given a follow-up dynamic CT scan that showed multiple, small, and variably-sized nodules in both hepatic lobes. These nodules appeared to be enhanced in the early arterial phase, and washed out in the portal phase (Figure 4). In addition, the lesions were stained by selective angiography of the hepatic artery (Figure 5), suggesting HCC. The patient refused liver biopsy. Considering her worsened hepatic functions, we performed a transcatheter arterial chemotherapy (TAC) with a modified dose of epirubicin and cisplatin. After the second course of TAC, a follow-up CT scan showed multiple densely lipodol-laden lesions that suggested HCC on the previous CT scan. Follow-up α-fetoprotein concentration also decreased to 43.7 ng/mL. However, her hepatic dysfunction became so severe that we stopped any further trans-arterial chemotherapy. Despite conservative treatment, she died of sepsis caused by aspiration pneumonia five months after the completion of the second TAC.
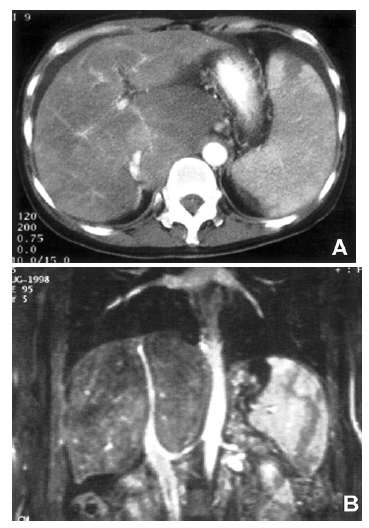
(A) CT scan showing narrowed intrahepatic vena cava, hypertrophied caudate lobe, mild ascites, and moderate splenomegaly with focal splenic infarction. (B) MR imaging showing obliterated hepatic veins and diffusely narrowed intrahepatic vena cava.
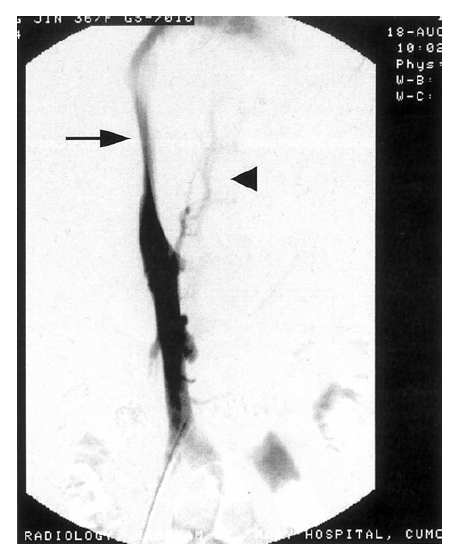
Venogram of the liver showing broad stenosis of the intrahepatic vena cava (arrow) with collateral vessels (arrow head).
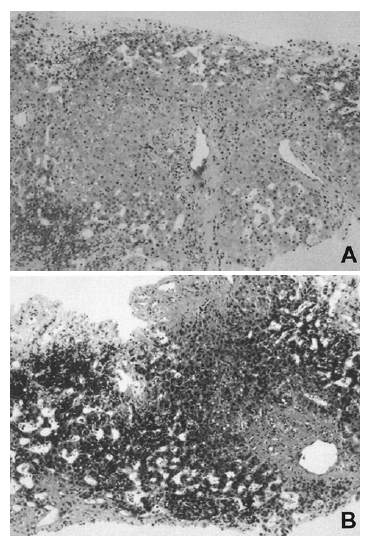
A and B. Histological findings of the liver showing zone 3 hemorrhage with fibrotic regenerative nodular formation (hematoxylin and eosin stain, ×100), and fibrotic expansion of portal area (Masson trichrome stain, ×100).

Liver dynamic CT scan showing multiple, small, and variably-sized nodules in both hepatic lobes. The nodules were enhanced in the early arterial phase (arrows; A), and washed out in the portal phase (arrows; B), suggesting hepatocellular carcinoma.

Selective angiography of the hepatic artery showing multiple, variably-sized, and hypervascular lesions in both hepatic lobes (arrows), indicating hepatocellular carcinoma.
DISCUSSION
BCS is a disease that causes the obstruction of the hepatic veins or the inferior vena cava (IVC). Its incidence is 0.019% among all autopsies: 0.015% in males and 0.023% in females3). With the help of newly developed imaging techniques, more cases of BCS are now being detected3, 4). However, BCS is still uncommon, and its pathogenesis, natural history, clinical features, and appropriate treatment are imperfectly understood.
BCS patients have been classified into various subgroups on the basis of their etiology, anatomy, histology, and clinical features3, 5, 6). Recently, a new classification of BCS was proposed by Okuda et al7). According to the new classification, BCS can be classified into two types: primary hepatic vein thrombosis and primary IVC obstruction. The latter is commonly called membranous obstruction of the IVC (MOVC), which is more frequently associated with HCC7). The majority of BCS cases in Japan3), Nepal8), China9), and South Africa10) are of the MOVC type. This is in contrast to the cases seen in the West11). Most cases of BCS in Korea are also of the MOVC type12), although there has been no detailed data on BCS in our country.
The severity of this disease depends on the extent of involvement of the hepatic veins or the IVC, and the development of collaterals. In most cases, primary hepatic vein obstruction presents in an acute or subacute form of BCS, whereas the MOVC type appears insidious or chronic7). However, the balance between the rate of progress of the underlying thrombotic process and the restorative thrombolytic process is important in the natural history of BCS6). Some prognostic factors related to the survival of patients with BCS have been investigated: the factors include age, response of ascites to diuretics, Child-Pugh score, serum creatinine, prothrombin time, serum aminotransferase, and treatment with porto-systemic shunting13, 14). Liver histopathologic findings did not show any relationship of the survival of patients with BCS in the study14).
BCS accounts for 0.7% of all cases of HCC4). Many cases of BCS-associated HCC have been reported with some geographic variation. Reports from Japan and South Africa show higher rates of association than those of other countries10, 15, 16), ranging from 6.4 to 47.5%3, 10, 15–18). These results suggest that many factors, such as chronic infection with hepatitis B or C viruses, may play a role in the development of HCC in patients with BCS17). However, HCC as a complication of BCS can be developed without underlying viral infections10, 18). The etiology of HCC arising in patients with MOVC has not been clearly identified. However, it is assumed that, due to hepatic venous outflow obstruction, hepatic congestion with extensive centrilobular necrosis and subsequent liver fibrosis can increase hepatic regenerative activity; thereby increasing DNA synthesis7, 19). This may play a part in hepatocarcinogenesis. Most cases of HCC arising from BCS are associated with MOVC, but they are rare in primary hepatic vein thrombosis. The former is milder and insidious for a longer period, whereas the latter is more severe, sometimes causing acute hepatic failure7). These clinical features, in part, seem to influence the differences in frequency of BCS-associated HCC between the two groups.
To the best of our knowledge, our case of HCC arising from BCS represents the most rapid progression yet reported. The mean reported intervals between BCS and HCC ranged between 4.5 and 24 years3, 18, 20). In this case, HCC developed only nine months after the diagnosis of BCS, and the patient died only eight months later. It was not clear why the clinical course of our patient was so severe and rapidly progressive. HCC reportedly develops more frequently in patients with primary IVC obstruction, and among these, broad obstruction of the IVC is mostly fatal3). While angiographically, our patient showed the mixed type, she mainly had the MOVC type. The pathological anatomy of an MOVC lesion varies in length from a thin membrane to a long stretch of the IVC, as seen in our patient7). Unfortunately, there are no comparative data on the rates of developing HCC according to the different lengths of lesions in patients with MOVC. As mentioned above, a long-segment narrowing of the IVC is eventually fatal, but it is unclear whether such an obstruction predisposes the progression to HCC from MOVC.
It is therefore likely that HCC in patients with BCS needs sufficient time to develop, and HCC, as a complication of BCS, occurs more frequently in patients with MOVC than in those with primary hepatic venous obstruction7). The latter usually presents an acute illness, so the likelihood of developing HCC is little. In our opinion, preventive management for the underlying hypercoagulable state in patients with hepatic venous thrombosis may in part contribute to the protection of the liver from further chronic injury and subsequent development of HCC.
Our patient had an unknown etiology for developing MOVC, similar to other reported types of MOVC. MOVC may be a congenital disease16), but Okuda et al.7) considered it more a consequence of organized thrombosis. However, it must take a long time for a thrombus to organize and form a membrane, even if it is an acquired disease. We think that prolonged chronic liver injury in this patient might have begun congenitally, or early in life. Meanwhile, the patient may have become more susceptible to environmental carcinogens as Kew et al. have assumed10). Moreover, she had been taking considerable amounts of medicinal slimming tea for more than five years with the intention of losing weight. Some unidentified chemicals in the tea may have worked as carcinogens, and this is worthy of further study.
HCC arising from BCS shows no clear trend, and its clinical course does not differ from that of HCC from other causes18). Because of multiple nodules, we decided to perform some cycles of TAC for treatment. However, her hepatic function became worse after two cycles of TAC with only 50% of the therapeutic dose, leading to her death. In retrospect, instead of shunt operation and TAC, liver transplantation would have been a better treatment for her before HCC developed.
In conclusion, we believe that patients with MOVC, especially those with long-segment narrowing of the IVC, should be carefully observed and followed closely as a high-risk group for developing HCC or a fatal group. More detailed investigations are necessary to characterize the types of HCC arising from BCS.