


INTRODUCTION
Multiple myeloma (MM) is a clonal B-cell malignancy that arises consistently from asymptomatic precursor conditions, specifically, monoclonal gammopathy of undetermined significance and smoldering MM. The proliferation of malignant plasma cells in the bone marrow and a subsequent overabundance of monoclonal paraprotein (M protein) in the serum and/or urine, renal dysfunction, anemia, hypercalcemia and lytic bone disease are the hallmarks of MM [1,2]. The incidence of the disease has increased rapidly in recent years, which, at least in Korea, accounts for it being one of the hematological malignancies currently in the medical and socioeconomic spotlight [3]. Despite significant advances in therapeutic strategies, including stem cell transplantation, proteasome inhibitors, and immunomodulatory drugs (iMiDs), which have led to improved responses to treatment and longer overall survival, most patients with MM eventually relapse and succumb to the disease [4,5]. Thus, there is a clear need to develop novel therapeutic options.
Recently, cellular immunotherapies have been recognized as an effective therapeutic modality for MM [6]. The human immune system has immense diversity and specificity that rely on a wide variety of effector mechanisms, such as those involving Fas ligands, the complement system, perforins, granzymes, and interferon-gamma (IFN-╬│) [7]. However, myeloma suppresses the immune response as a whole by releasing immune suppressive molecules and cytokines, leading to the tumorŌĆÖs escape from the effector immune response [8]. The goal of cancer immunotherapy is to activate, restore, and augment cytotoxic effector cells at the tumor site to effectively kill the tumor, all of which rely on the safe induction of cytotoxic cells that recognize and kill tumor cells [9]. An ideal immunotherapy should overcome the effects of an immunosuppressive microenvironment, train and recruit immune cells to eliminate all cancer cells, improve patient outcome without affecting healthy cells, and remain active in the event of recurrence. Dendritic cell (DC) vaccination and adoptive cell immunotherapy with chimeric antigen receptor (CAR) T-cells, T-cell receptor (TCR)-engineered T-cells, and natural killer (NK) cells are emerging as promising forms of cellular immunotherapy in patients with MM [10-15]. This review focuses on the efficacy and safety of recent preclinical and clinical trials in the development of DC vaccines, genetically engineered effector T-cells, and NK cell therapies for MM.
DENDRITIC CELL VACCINATION
The most potent antigen-presenting cells are DCs, which play a vital role in recognizing, processing, and presenting antigens on the cell surface of naïve T-cells, and modulating tumor specific immunity [16,17]. In MM, the functional ability of DCs is abolished by several immunosuppressive cytokines and inhibitory proteins, such as vascular endothelial growth factor, interleukin 10 (IL-10), IL-6, and transforming growth factors (e.g., TGF-β), secreted by malignant plasma cells and the tumor microenvironment [18]. Thus, the retrieval of fully functional DCs against tumor cells is a promising therapeutic strategy. Among the factors that need to be considered for successful DC vaccination strategies are the selection and source of the tumor antigen, the potency of the DC vaccine formulation, the mode of delivery, adjuvants, and immunomodulation, and the treatment schedule. In general, DCs generated ex vivo for cancer immunotherapy should be mature, capable of migrating in the direction of secondary lymphoid organs, and produce type 1 helper (Th1) polarizing cytokines. In a previous study, we reported that functionally active DCs generated ex vivo from patients with MM exhibited the properties of the strong, mature DCs necessary to induce potent myeloma-specific cytotoxic T lymphocytes (CTLs) [13,19].
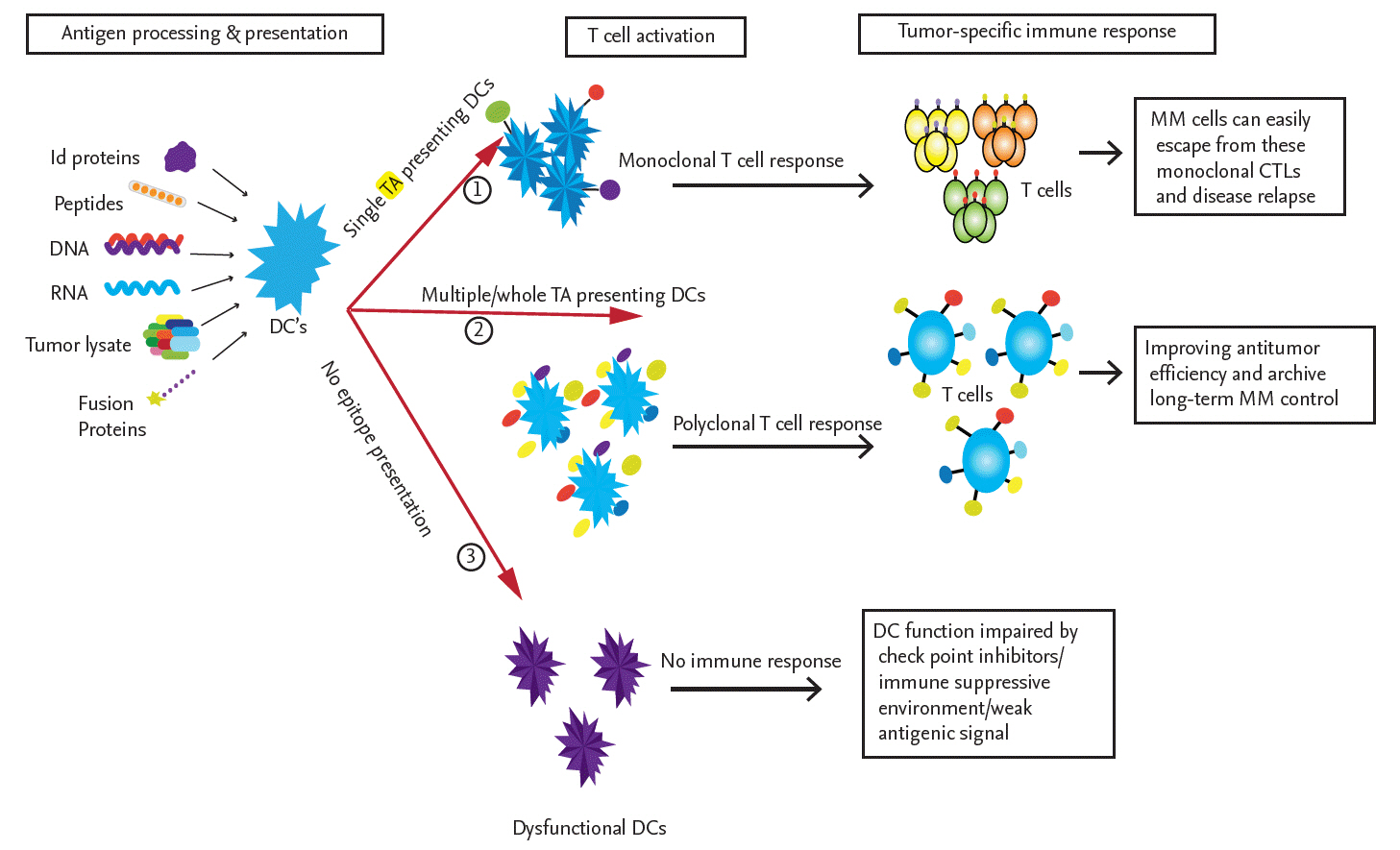
In early clinical trials of immunoglobulin idiotype (Id)-pulsed DCs, features indicative of myeloma- specific immune responses were observed but the clinical responses were unsatisfactory because of the weak antigenicity of the Id [20]. Tumor-associated antigens (TAAs)-loaded DCs may also induce tumor-specific CTL responses for targeting myeloma cells in vitro. Various TAAs have been identified in MM, such as mucin 1 (MUC1), New York esophageal squamous cell carcinoma 1 (NY-ESO-1), and telomerase reverse transcriptase (hTERT). However, although a single TAA may induce an antitumor immune response in MM, the myeloma cells can escape immune recognition via the down-regulation of this specific antigen over time. To overcome this problem, DCs can be loaded with whole myeloma cells to improve the antitumor immune response effectively and avoid tumor cell immune escape [13,19]. This alternative approach has been tested using DCs loaded with myeloma cell lysates or apoptotic bodies, DCs transfected with tumor-derived RNA or heat shock proteins (HSP) gp96, and DC-myeloma fusions. Regardless of the specific method, the results showed that multiple unknown epitopes were presented by the DCs for major histocompatibility complex (MHC) I recognition and the subsequent induction of polyclonal T-cell immune responses to effectively kill myeloma cells [20]. Our group was also able to generate potent DCs loaded with dying myeloma cells, which induced myeloma-specific CTLs with strong Th1 polarization [21-27]. In our previous phase I/IIa study of patients with relapsed or refractory MM, DC vaccination using cells loaded with ╬│-irradiated dying myeloma cells was well tolerated, did not result in any significant adverse effects, and led to disease-stabilizing activity in 66.7% of the patients and a 77.8% immunological response [28]. Fig. 1 describes DCs pulsed or loaded with different sources of myeloma antigens to induce myeloma-specific immune responses.
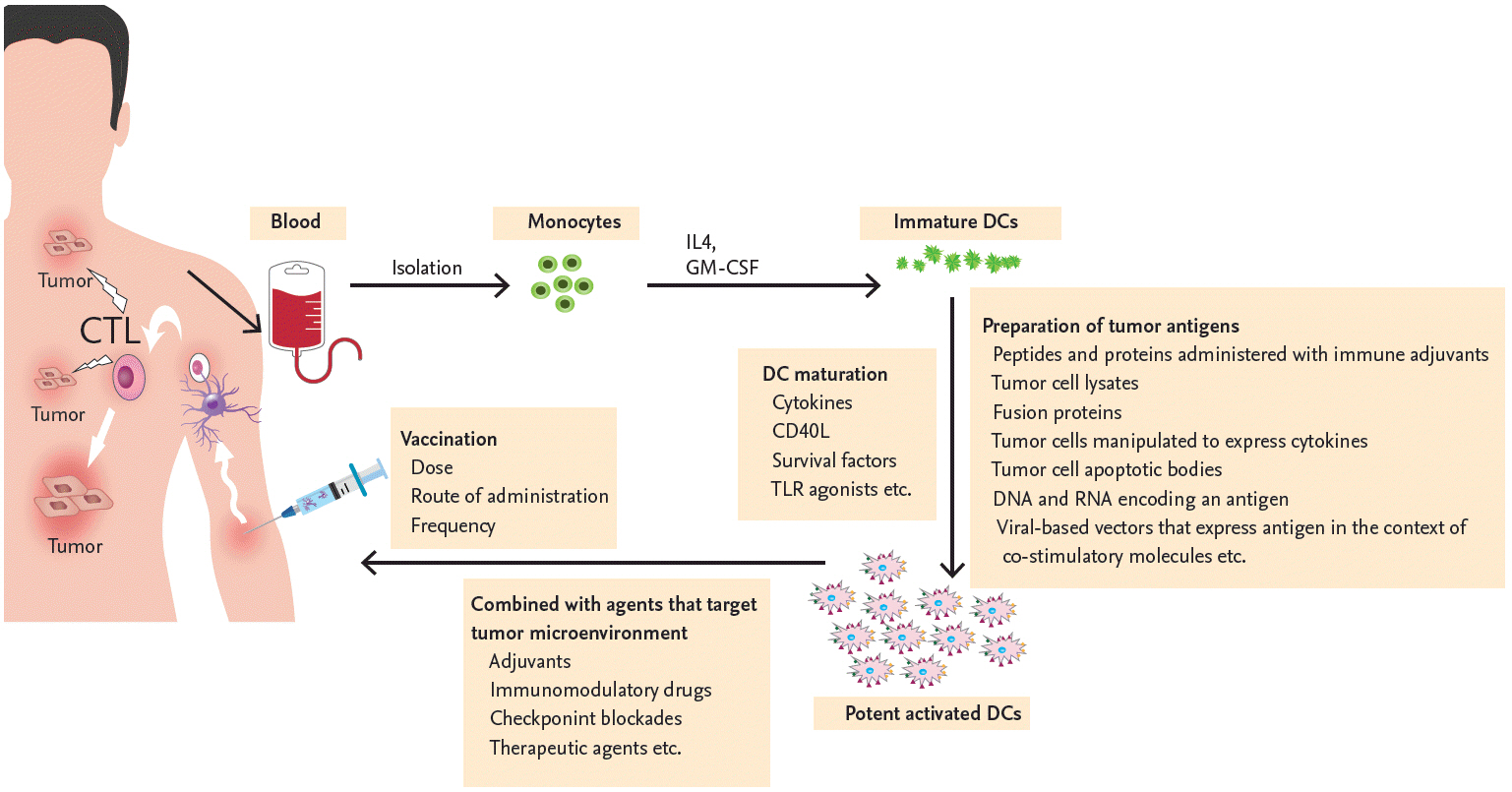
Recent attempts to improve the effectiveness of DC vaccines have included the use of a cocktail of several tumor antigens, genetic engineering and molecular biological modifications, and combinations with other agents. IMiDs, such as thalidomide, lenalidomide, and pomalidomide, were also shown to be effective in patients with MM. The immunological mechanism of iMiDs involves the down-regulation of regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and an enhancement of the immune response by activating NK cells and T-cells [29]. We previously reported a strong synergistic effect of DC vaccination and lenalidomide [30] or pomalidomide [31] in the induction of an anti-myeloma effect in a murine myeloma model. Another method to improve DC vaccination is to combine checkpoint blockades that modulate negative regulation in the tumor microenvironment [32]. Our data, obtained in a murine myeloma model, indicated a remarkable anti-myeloma effect of DC vaccination when combined with an antiprogrammed cell death 1 (PD-1) antibody and lenalidomide, attributable to an augmented immune response [33]. These improvements in DC vaccination may give rise to a promising cell therapy tool able to induce myeloma-specific immune responses, without significant adverse effects. A schematic representation of the future perspectives of enhanced DC vaccination strategies is shown in Fig. 2.
GENETICALLY ENGINEERED T-CELL THERAPY
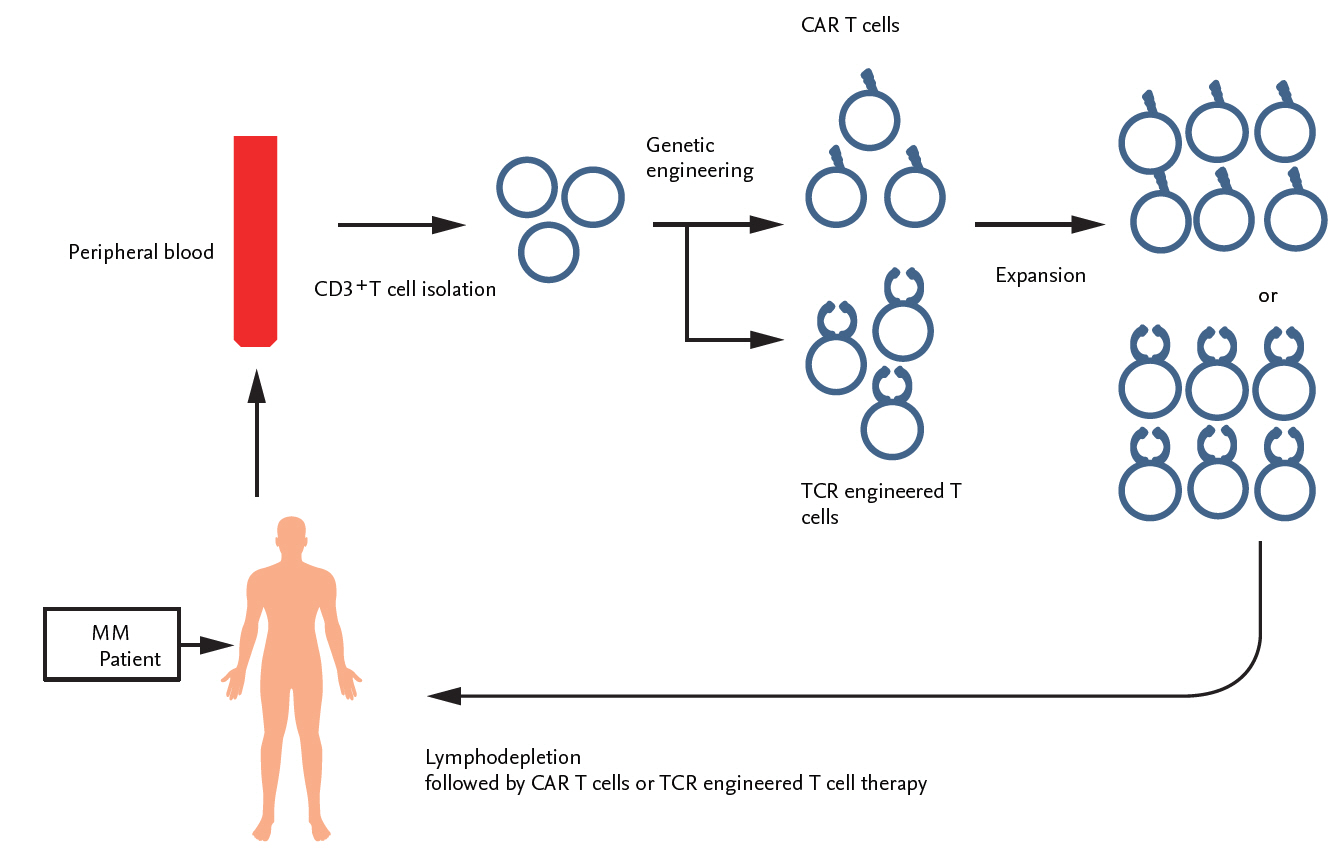
Approaches aimed at triggering a tumor-specific T-cell response and, thus, immunological memory against the tumor cells, include the adoptive transfer of genetically engineered T-cells. This is achieved by introducing antibody-like recognition in CARs or by modifying TCR specificity. Both methods should result in the targeting of surface antigens that are highly expressed in MM. A schematic representation of the treatment of MM with genetically engineered T-cells is shown in Fig. 3.
CAR T-cell therapy
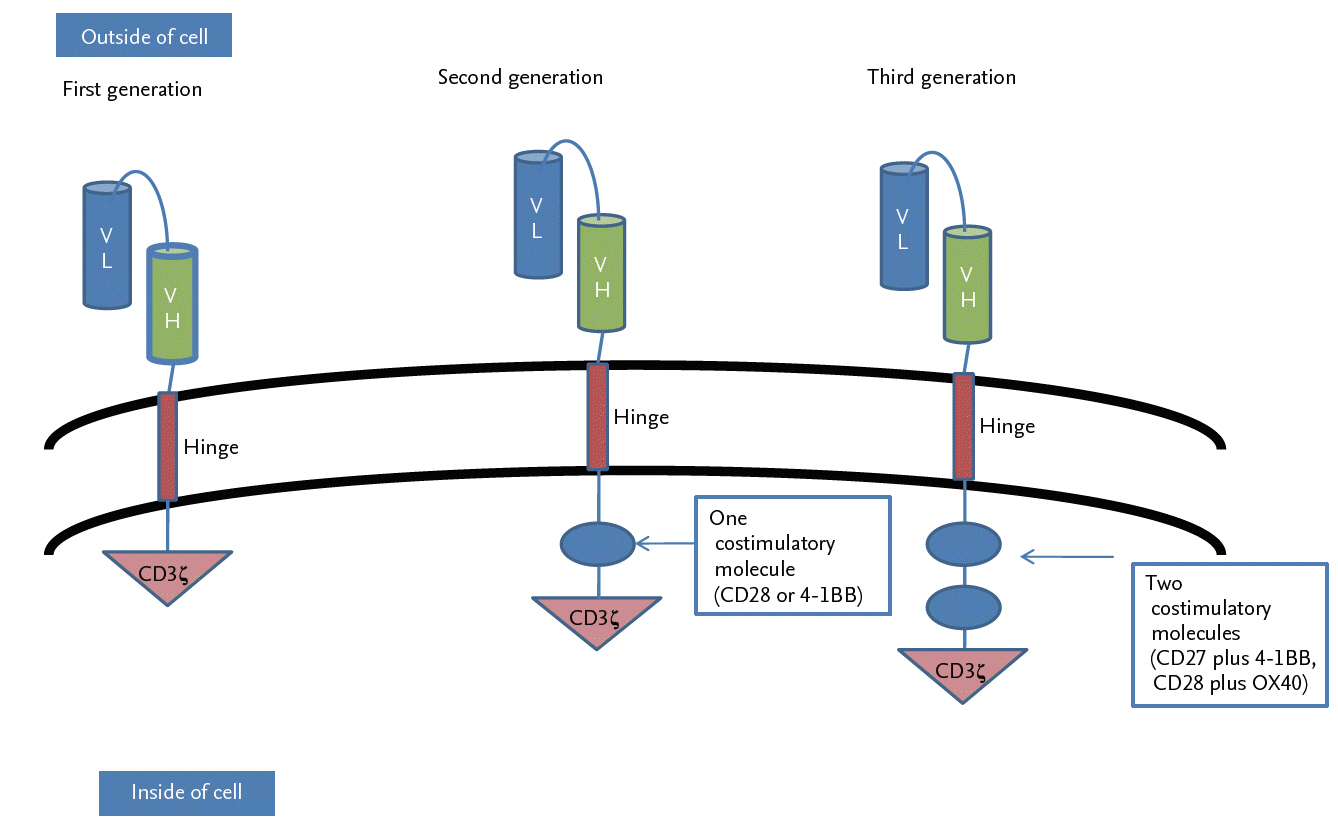
CAR T-cells are genetically engineered T-cells that can recognize specific antigens expressed on tumor cells and then kill the tumor cells [34,35]. A CAR consists of three domains: a single chain variable fragment (scFv) linked to a transmembrane domain, costimulatory domains, and a T-cell activation domain [36]. First-generation CAR T-cells contained only a single signaling unit, derived from the cluster of differentiation 3╬Č (CD3╬Č) chain or ╬│ chains of the high-affinity IgE receptor (Fc╬ĄRI╬│), as an intracellular signaling domain. However, due to their restricted cytokine secretion and T-cell production, both types showed very weak antitumor activity in the killing of tumor cells [37]. Further evolutions of CARs improved their therapeutic safety and efficacy by adding one or more costimulatory molecules. Thus, second-generation CARs had a single costimulatory domain derived from either CD28 or TNF receptor superfamily member 9 (4-1BB), and third-generation CARs had two costimulatory domains, such as CD27 plus 4-1BB or CD28 plus tumor necrosis factor receptor superfamily, member 4 (OX40). (Fig. 4) [38].
The first gene-modified CAR T-cell therapy, formerly known as CTL019, yielded a remarkable response in patients with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL), resulting in approval of this therapeutic approach in the United States [39]. The excellent outcome of anti-CD19 CAR T-cell therapy against B-ALL motivated the development of myeloma cell-specific CAR T-cells. Requirements for the target antigen, a crucial factor in CAR development, were that it was expressed uniformly and specifically on all malignant cells (on-target) but, to avoid toxicity, not by normal tissues (off-target). Several antigens have been studied as feasible myeloma targets for anti-myeloma CAR T-cells, including CD44 variant 6, CD70, CD56, CD38, CD138, CD19, immunoglobulin kappa light chain, signaling lymphocytic activation molecule F7 (SLAMF7), and B-cell maturation antigen (BCMA) [40]. All have their limitations in terms of their use in MM. CD44 variant 6 is equally expressed on activated T-cells and monocytes [41], CD70 on activated lymphoid cells [42], and CD56 on NK cells, T-cells, and neuronal cells [43]. Although CD38 is normally expressed on precursor B-cells, plasma cells, T-cells, NK cells, and other tissue cells, it is highly expressed on malignant plasma cells, which has motivated the development of anti-CD38 CAR T-cells [44,45]. CD138 is also expressed on plasma cells and several tissue cells but it is nonetheless a good candidate target on myeloma cells [46,47]. The anti-CD19 CAR T-cells used in B-ALL showed impressive results against myeloma and may deplete myeloma stem cells, despite the minimal expression of CD19 on myeloma cells [48]. Immunoglobulin kappa light chain is expressed by mature B-cells but may be a target for the MM stem cell population expressing these surface immunoglobulins [49]. SLAMF7 is a promising target because of its strong expression on myeloma cells, despite its expression on plasma cells, NK cells, CD8+ T-cells, monocytes, B-cells, and DCs. Among these antigens, BCMA is currently being tested in several clinical trials and the preliminary results have been impressive. BCMA is uniformly expressed on all MM cells but not on hematopoietic stem cells or other immune cells [36].
Currently, four clinical trials of CAR T-cell therapy targeting BCMA are ongoing in patients with relapsed or refractory MM (Table 1). Raje et al. [50] presented the updated results of a multicenter study of bb2121 anti-BCMA CAR T-cell therapy in patients with heavily pretreated MM. The 43 patients received 50ŌĆō800 ├Ś 106 CAR T-cells after lymphodepletion with cyclophosphamide and fludarabine. A dose-escalation study showed that a minimum of 150 ├Ś 106 CAR T-cells were needed to achieve an optimal outcome. An assessment of the overall response rate (ORR) showed that 77% of the patients had a complete response (CR), including 44% with a stringent CR (sCR), with comparable rates in patients with high- and low-BCMA-expressing tumors. Median progression-free survival following treatment with Ōēź 150 ├Ś 106 cells was 11.8 months. In addition, bb2121 CAR T-cell therapy was relatively well tolerated. Cytokine release syndrome (CRS) of any grade developed in 63% of the patients, but in all cases it was manageable. Safety and efficacy data for BCMA-specific CAR T-cells (CART-BCMA) in the treatment of refractory MM were also reported [51]. CART-BCMA had activity in patients with heavily pre-treated MM, with an ORR of 46%, and its activity was not clearly associated with baseline BCMA expression or soluble BCMA concentration. The main non-hematological toxicities associated with CART-BCMA were CRS and neurotoxicity. CRS of any grade developed in 83% of the patients and Ōēź grade 3 CRS in 29%. Smith et al. [52] presented the results of a phase I study of MCARH171 (human scFv-derived BCMA-targeted CAR T-cells) in patients with relapsed or refractory MM. Six patients who received 0.7ŌĆō8 ├Ś 108 CAR T-cells after undergoing lymphodepletion with cyclophosphamide and fludarabine therapy experienced manageable CRS; there was no case of CRS Ōēź grade 3. In addition, Chinese investigators reported the results achieved with LCAR-B38, which targets BCMA [53]. Forty pat ients who had received at least three prior lines of therapy and whose malignant plasma cells had > 10% BCMA expression were included in the study. The ORR in 22 evaluable patients was a striking 100%, with 64% of patients achieving sCR. CRS of any grade was seen in 85% and CRS Ōēź grade 3 in 8.6%. Finally, the results of an interesting study of the combined infusion of CD19- and BCMA-specific CAR T-cells in patients with relapsed or refractory MM were recently reported [54]. A deep and long-term remission in patients treated with both CD19 CAR T-cells and BCMA CAR T-cells was anticipated, because of the association of disease relapse with myeloma stem cells. Nine of the 10 evaluable patients achieved a partial response or better. CRS occurred in all patients. Long-term follow-up results are needed to determine whether this combined approach improves outcomes.
T-cell receptor-engineered T-cells
TCRs are expressed on the surface of T-cells and recognize both intracellular and extracellular antigens. However, many tumor cells down-regulate the expression of MHC molecules to escape from immune cells. TCRs recognize and bind to peptides loaded onto MHCs, resulting in the activation of several signaling cascades and, in turn, protein phosphorylation. Among the proteins involved in these signaling cascades are nuclear factor of activated T-cell (NFAT) proteins and nuclear factor Fos. Their activation results in that of T-cells and, thus, the release of IFN-╬│, granzyme B, and IL-2 [55]. TCR-modified T-cells are engineered to encode receptors for the tumor antigen peptide-MHC complex. Genetically engineered TCR are established by modifying the specificity of the ╬▒ and ╬▓ chains of the TCR to a particular tumor antigen for enhanced antigen recognition [47].
Currently, engineered TCR therapies mainly focus on cancer testis antigens (CTAs), which are highly expressed by tumors and expressed only by male germ cells in the testis and placenta but not in adult normal tissues [56]. Thus, engineered TCRs targeting CTAs are a promising therapy for cancer [56,57]. TCR-engineered T-cells targeting CTAs loaded onto human leukocyte antigen (HLA)-A have been employed in the treatment of MM. A phase I/II study of NY-ESO-1 targeting engineered TCRs was conducted in 20 patients with advanced MM and sarcoma. The results showed a significant clinical response and the remarkable safety of this approach, as none of the patients experienced CRS or other infusion-related toxicities. Recent innovative clinical trials to develop engineered TCRs against MM have focused on CTAs, neoantigens, and TAAs, such as melanoma antigen recognized by T-cells 1 (MART1), MAGE family member A3 (MAGE A3), and L antigen family member 1 (LAGE-1) [57,58].
NATURAL KILLER CELLS
NK cells are a subset of peripheral blood lymphocytes characterized as effector cells of the innate immune system, with potent cytotoxic activity toward cancer cells or infected cells [59]. Target-cell killing by NK cells occurs via perforin/granzymes granule-mediated lysis and death receptor interaction. NK cells also lyse target cells coated with antibodies to the antigen on the tumor cell surface, by antibody-dependent cellular cytotoxicity (ADCC). Target cell recognition is mediated by the signals delivered through several activating and inhibitory receptors of NK cells, including natural killer G2D (NKG2D), DNAX accessory molecule-1 (DNAM), and natural cytotoxicity receptors such as NKp30, NKp44, NKp46 as activating receptors, and killer-cell immunoglobulin-like receptors (KIRs), heterodimeric C-type lectin receptor (NKG2A/CD94), and check point T cell immunoreceptor with Ig and ITIM domains (TIGIT) as inhibitory receptors [60]. The advantage of NK cell therapy is that it results in the killing of various target cancer cells without prior sensitization. It also does not cause graft versus host disease (GVHD) [61]
Myeloma cells evade host immunity through various mechanisms, such as the activation of tumor suppressive pathways and suppressive cytokines (i.e., IL-6, IL-10). Among the involved immune cells are NK cells, which become weak or dysfunctional within the MM micro-environment. In patients with advanced MM, the number of circulating NK cells is reduced and their function is suppressed [62]. Advances in the ex vivo activation and expansion of NK cells to obtain adequate cell numbers have been reported, including the demonstration of an anti-MM effect of NK cells [63-65]. In attempts to develop NK cell therapies for MM, although the clinical safety of autologous ex vivo expanded NK cells infusion in patients with MM has been shown, remarkable clinical outcomes have not been achieved [66]. In contrast, allogeneic NK cells [60,61] treated with proteasome inhibitors and other anti-myeloma drugs, such as bortezomib or iMiDs, were recently shown to significantly induce NK cell effector function by inducing the expression of NK activating receptors [6]. A phase I trial evaluated the use of cord blood (CB)-derived ex vivo expanded NK cells as part of a conditioning regimen with high-dose melphalan and lenalidomide before autologous stem cell transplantation in 12 patients with MM, with high-risk characteristics. Ten of the patients achieved, at least, a very good partial response, including eight with a CR, without any significant infusion toxicities or GVHD. Interestingly, CB-NK cells were detected as an activated phenotype (NKG2D+/NKp30+) in vivo until day 21 after autologous stem cell transplantation [67]. There are several ongoing trials of NK cell therapy aimed at enhancing activity against MM, such as the use of NK cells in combination therapy with iMiDs including lenalidomide and pomalidomide, checkpoint inhibitors [68], and monoclonal antibodies to improve clinical efficiency (Fig. 5) [64,68].
Analogous to CAR T-cells, several new NK-based approaches, such as the use of NK cell lines (i.e., NK-92-CS1-CAR cells) [69], other genetic engineering approaches [60], and the generation CAR NK cells specifically targeting MM cells [70,71], have been developed. In preclinical models, CAR-engineered NK cells specific to SLAMF7 showed anti-MM activity [69], and bispecific forms redirecting NK cells to tumors have been developed for other hematological cancers [72]. With the innovative techniques in NK cell biology that are continually being developed, the prospects of novel, effective NK cell-based therapies for MM are eagerly anticipated.
CONCLUSIONS
Cellular immunotherapy offers great hope for the treatment of myeloma, given the encouraging results of preclinical and clinical trials. Cancer immunotherapy using DCs, NK cells, and genetically engineered T-cells has shown promise for the treatment of MM. The rapidly developing technologies support advanced strategies, including those relying on gene modification of effector cells, anti-myeloma drugs, and antibodies. These cellular immunotherapies are expected to play an important role in improving the outcome of patients with MM.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





