


To the Editor,
Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant inherent disorders with an incidence of one in 3,000 individuals [1]. NF1 can affect the whole body including the neurological, cardiovascular, gastrointestinal, endocrine, and orthopedic systems. Most patients with NF1 experience multiple neurofibromas, skin fold freckles, caf├®-au-lait spots, and Lisch nodules, usually before puberty. Furthermore, a higher overall risk of malignancy is reported in patients with NF1 than the general population. Here, we report a novel germline mutation in the NF1 gene in a patient with NF1 and pheochromocytoma.
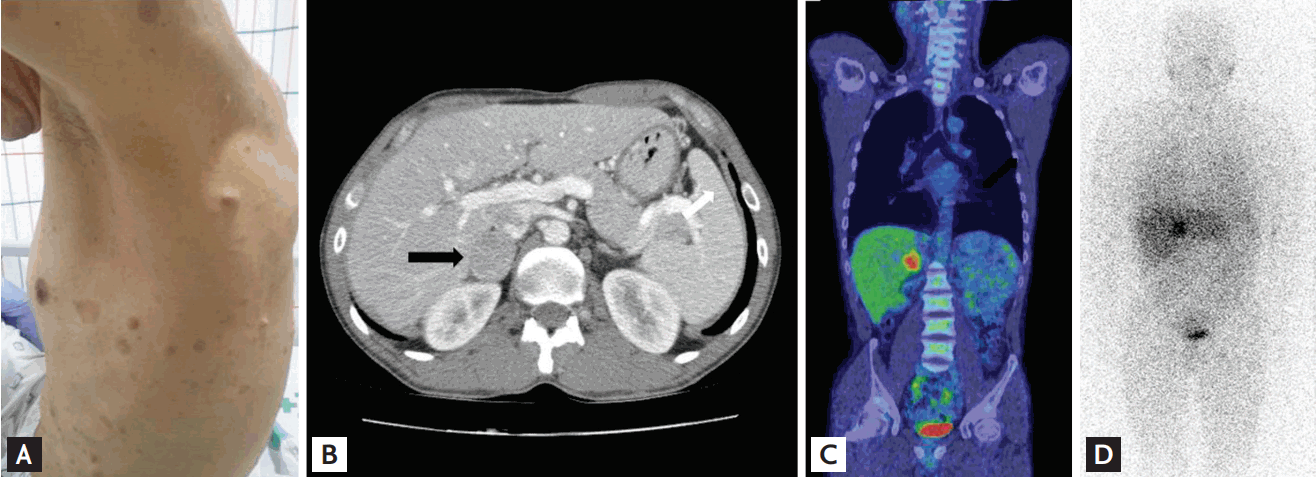
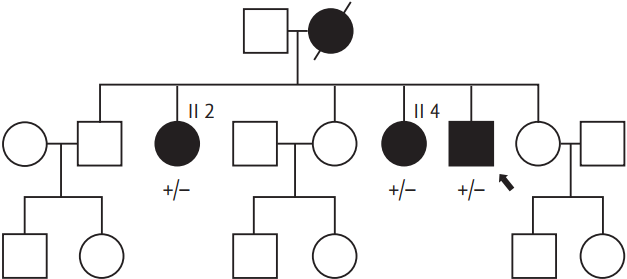
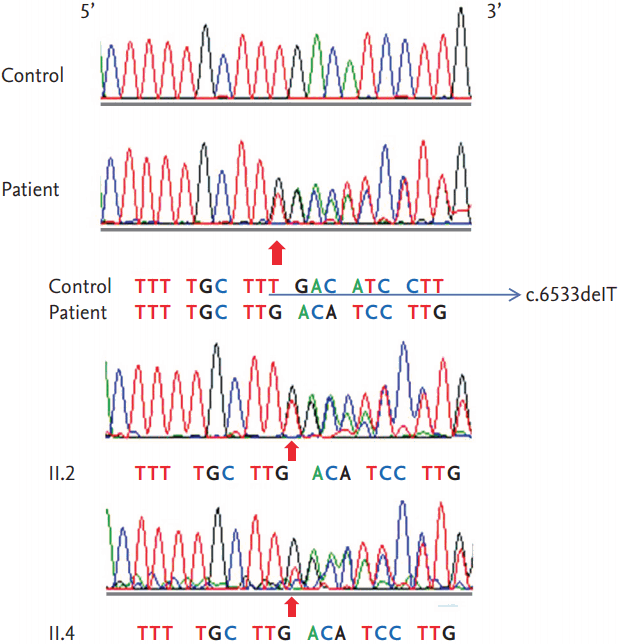
A 35-year-old man was referred to Incheon St. Mary's Hospital, The Catholic University of Korea, because of recurrent duodenal ulcer bleeding. He had surgery for duodenal tumor removal about 5 years ago, but there was no pathologic record available. A few months prior, the patient had visited a private clinic for gastrointestinal bleeding and an endoscopy identified diffuse oozing from the ulcer at the descending portion of the duodenum. It was revealed that this was treated elsewhere with hemoclips in an endoscopy unit; there was also stenosis and deformity at the duodenal bulb. The patient was treated with parenteral nutrition and a proton pump inhibitor but recurrent bleeding still occurred. Abdominal computed tomography (CT) performed at a private clinic revealed a large intramural hematoma at the descending duodenum and a mass was found on the right adrenal gland. In Incheon St. Mary's Hospital, The Catholic University of Korea, , an endoscopic examination showed a 0.4 ├Ś 0.3 cm sized A1 ulcer at the superior duodenal angle with sign of recent hemorrhage (Forrest IIa). Although bleeding was stabilized and a follow-up abdominal CT showed complete regression of the duodenal hematoma, a heterogeneously enhancing mass in the right adrenal gland (3.3 ├Ś 2.6 cm) and high attenuation value was still noted (> 20 Hounsfield unit) (Fig. 1A). As a result, the patient was referred to the Endocrinology Department. Upon physical examination, it was found that he had multiple caf├®au-lait spots, axillary freckling, and many cutaneous neurofibromas (Fig. 1B). Within a decade, he had received multiple resections for the subcutaneous masses that were dispersed on the scalp and the inguinal areas and were histologically reported as neurofibromas. However, no Lisch nodules or optic gliomas were found by ophthalmologic examination. The patient was compatible with NF1 based on the National Institutes of Health criteria [2]. His height was 160 cm and body weight 59 kg and he exhibited normal development and mental function without neuromuscular deformities. His blood pressure had been 120/80 mmHg without antihypertensive agents and there was no reporting of palpitation, diaphoresis, excessive sweating, or headache. Renin plasma activity was 0.72 ng/mL/hr, aldosterone was 1.7 ng/dL, indicating normal aldosterone-to-renin ratio (9.6). The serum cortisol after 1 mg dexamethasone was normally suppressed (0.64 ╬╝g/dL). There were increases in serum norepinephrine (842.1 pg/mL; normal, 100 to 410) and urinary normetanephrine (380.79 ╬╝g/day; normal, < 80), metanephrine (5,175.2 ╬╝g/day; normal, 88 to 444), and vanillylmandelic acid (20.75 mg/day; normal, < 8), which was indicative of pheochromocytoma. Fluorine-18 fluorodeoxyglucose- positron emission tomography and 123I-metaiodobenzylguanidine scintigraphy confirmed a localized increased uptake in the right adrenal gland (Fig. 1C and 1D). A laparoscopic right adrenalectomy and pathologic report was also consistent with pheochromocytoma. To detect a possible mutation in NF1, we performed a genetic analysis of the patient and his two sisters who showed clinical signs of NF1 (Fig. 2). Genetic investigations were conducted with informed consent from the proband and two sisters who showed skin manifestations suspicious of NF1. The Ethics Committee of Incheon St. Mary's Hospital approved the research protocol. Genomic DNA was isolated from peripheral white blood cells using a Flexigene DNA kit (Qiagen, Hilden, Germany). Polymerase chain reaction (PCR) was performed using intronic primers for the 60 exons of the NF1 gene. Purified PCR products were sequenced with an ABI3730xl genetic analyzer (Applied Biosystems, Foster City, CA, USA) and analyzed using a Finch TV version 1.2.0 (Geospiza, Seattle, WA, USA). All nucleotide numbers refer to the wild-type genomic DNA sequence of the NF1 gene as logged at the National Center for Biotechnology Information. Mutations were identified by comparing with the reported cDNA reference sequence (GenBank accession number: NM_000267.3). Genetic analysis of the index patient detected a novel germline NF1c.6533delT in exon 43 of the NF1 gene, and the identical germline mutation was found in both sisters (other family members refused genetic analysis) (Fig. 3). This mutation resulted in a frame shift with shortening by 641 amino acids (p.L2178XfsX641). No reports were found at this position in the National Center for Biotechnology Information and none of these novel sequence variations were found in 23 unrelated Koreans.
NF1 is a large gene that contains 57 constitutive exons, three alternatively spliced exons, and spans approximately 350 kb of genomic DNA [1]. NF1 encodes neurofibromin that acts as a GTPase-activating protein and inhibits the RAS (renin-angiotensin system) system that plays a major role in tumor formation. This is caused by a mutation in the NF1 gene and results in a predisposition to cancer with rates of malignancy 5% to 15% higher than the general population; primarily due to markedly increased risks of malignant peripheral nerve sheath tumors and central nervous system tumors including optic glioma [3]. The most common cause of death in NF1 patients is malignancy, with an average reduction in life expectancy of 8 to 15 years. Gastrointestinal stromal tumor (GIST), rhabdomyosarcoma, somatostatinoma, breast cancer, and pheochromocytoma are also known as common tumors in NF1 patients.
Our patient was also affected with pheochromocytoma, which affects 1% to 5% of patients with NF1; approximately 10 times greater than the general population [1]. Thus, physicians should be mindful of pheochromocytoma, especially if the patient is hypertensive. In addition, the patient reported a history of duodenal tumor resection and the literature reports that gastrointestinal tract involvement occurs in 12% to 15% of NF1 patients but with only 5% being symptomatic [4]. The most common types of tumors include neurofibromas, GIST of the small bowel, and periampullary somatostatinoma. Other tumors, including adenocarcinomas and neuroendocrine tumors (e.g., carcinoid tumors and gastrinoma) have also been reported in patients with NF1. Unfortunately, we could not have access to the pathologic reports of the duodenal mass that had been surgically removed from the current patient.
Until now, there has been limited information regarding a genetic-clinical association except that the deletion of a whole NF1 gene (manifesting early, large numbers of cutaneous neurofibromas, severe cognitive abnormalities, somatic overgrowth, and facial dysmorphism) and 3-bp in-frame deletion of Exon 17, c.2970ŌĆō2972 delAAT (manifesting typical pigmentary features of NF1, but no cutaneous or surface plexiform neurofibromas) [5]. In addition, genetic analysis of NF1 remains difficult because of its large size, lack of hot spots, inter- and intra- familial variability in disease expression, and more than 50% of de novo mutation. In the case presented here, we were unable to determine whether the present novel mutation in the NF1 correlates with pheochromocytoma as the sisters of the patient showed no signs of pheochromocytoma and refused further evaluation. Therefore, further studies are required to determine the clinical application of genetic information, which has the potential to result in better patient treatment and genetic counseling of family members. In conclusion, we identified a novel NF1c.6533delT mutation in a Korean patient with NF1 and pheochromocytoma.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





