


To the Editor,
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary cystic disease of the kidney. Renal cell carcinoma (RCC) is more frequently found in patients with ADPKD than in the general population, showing the prevalence of 5.3% to 8.3% in ADPKD patients with chronic kidney disease (CKD) [1]. The direct association between ADPKD and RCC development is difficult to speculate, and a certain proportion of reported cases of RCC in ADPKD patients with advanced CKD might be related to acquired cystic kidney disease. A few characteristic features can be found in RCC in patients with ADPKD: (1) early age at diagnosis (45 to 50 years vs. 61 to 62 years in the general population); (2) bilateral and multicentric occurrence; and (3) high frequency of papillary type especially among predialysis patients or the coexistence of two different subtypes [1-3]. A greater proportion of ADPKD patients have papillary-type RCC, since these phenomena have pathogenetic similarity to hyperplasia of the epithelial cells lining the cyst, which frequently form papillary-like structures in early pathologic studies. Analogous to inherited cancers, a two-hit hypothesis was proposed to explain the focal nature of renal cysts in ADPKD. However, the molecular genetic mechanisms of RCC in ADPKD have not been reported. Herein, we report an ADPKD patient with preserved renal function and a cystic tumor that showed aggressive progression after a long indolent clinical course. We suggest Xp11.2 translocation as a possible mechanism of RCC in ADPKD.
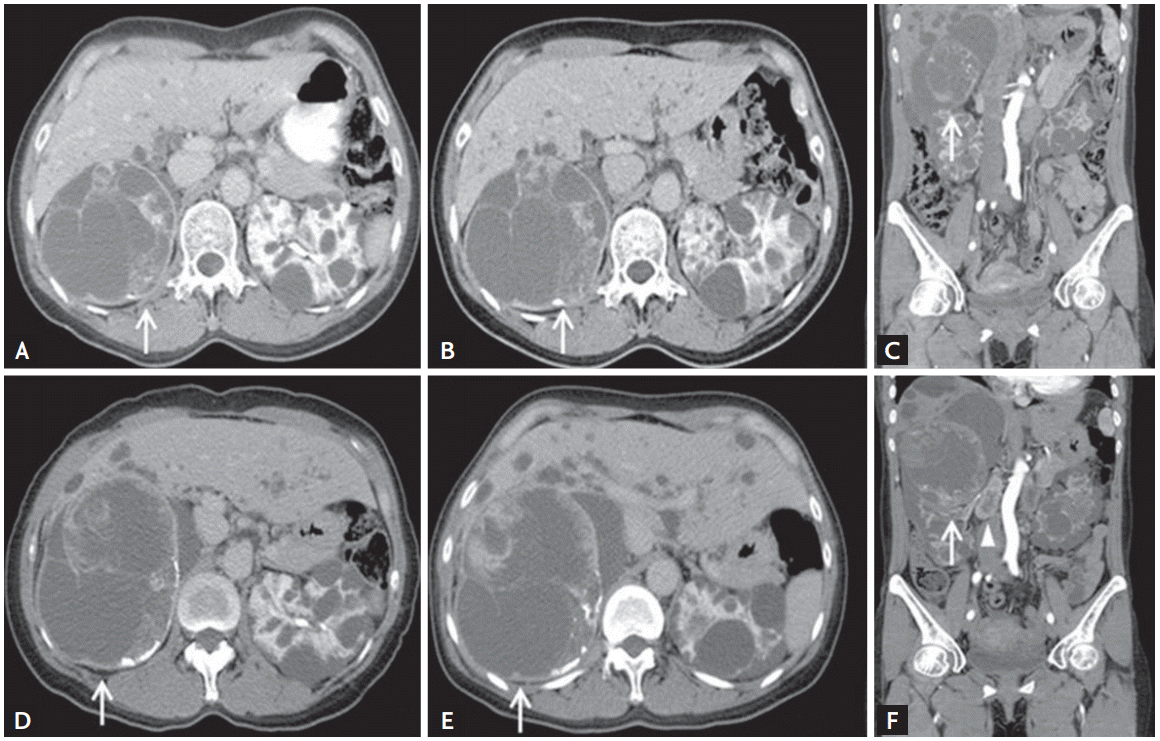
A 40-year-old woman presented with an enlarging cystic mass in the right kidney. At the age of 30, she was diagnosed with ADPKD characterized by innumerable, variable-sized cysts in both kidneys and liver despite no family history of ADPKD. At the age of 40, during her first visit to our hospital 10 years ago, a 10 ├Ś 11-cm cystic lesion with inner septation and a well-enhancing solid portion was detected in the right kidney upper pole by abdominal computed tomography (CT) (Fig. 1A). She chose not to undergo nephrectomy because of her young age and possible renal function deterioration. The cystic mass lesion was followed closely with biennial radiologic evaluations. Serial imaging studies showed no significant changes in size or radiologic features for 5 years (Fig. 1B and 1C). After 3 years, the size of the cystic mass started to increase to 10 ├Ś 12 cm, and enlarged lymph nodes were detected in the retrocaval and aortocaval areas (Fig. 1D). Since the size of the cystic mass further increased to 15.3 ├Ś 13 ├Ś 11 cm accompanied by extensive necrosis (Fig. 1E and 1F), she was admitted for right radical nephrectomy.
The patient had no hematuria or pain related to the kidney and was free from systemic complications including fever, anorexia, fatigue, and weight loss. Her hypertension was controlled with an angiotensin receptor blocker, and her serum creatinine level was 1.08 mg/dL. Cyst aspiration and cytologic examination performed 6 months prior to the last admission did not show any malignant cells. A polycystic kidney with a tan-yellow tumor accompanied by necrosis and hemorrhage was intraoperatively observed (Fig. 2A and 2B). Microscopic examination revealed RCC containing clear cell components accompanied by diffuse papillary architecture (Fig. 2C). Transcription factor E3 (TFE3) was strongly expressed, and the patient was positive for CD10 and negative for CK7 (cytokeratin 7) expressions, as measured by immunohistochemistry. Fluorescence in situ hybridization confirmed Xp11.2 translocation by showing a breakapart signal in TFE3 (Fig. 2D). We performed targeted exome sequencing for polycystic kidney disease 1 (PKD1) and PKD2 using whole blood but could not detect any mutations. After discharge, multiple metastatic lesions were detected in both lungs. Currently, she is receiving chemotherapy including a tyrosine kinase inhibitor (Pazopanib).
Our case is characterized by normal renal function and an absence of risk factors for RCC, except hypertension. The cystic renal mass was identified at a relatively young age and had not progressed over 5 years. However, clinical clues such as regional metastases and biologic aggressiveness, including vascular invasion and a high nuclear grade detected via pathology, allowed us to rule out more common and benign subtypes such as clear cell papillary RCC or multilocular cystic RCC. Immunohistochemical and cytogenetic examination confirmed Xp11.2 translocation in our case, which is the first report in an adult patient with ADPKD.
Xp11.2 translocation RCC has been increasingly reported in adults, accounting for 1.6% to 5% of adult RCC [4]. Compared to pediatric cases, Xp11.2 translocation RCC in adults shows an aggressive clinical course, showing an advanced stage with distant metastasis and poor prognosis. Xp11.2 translocation causes overexpression of various proteins such as ASPL (alveolar soft part sarcoma)-TFE3, PSF (PTB-associated splicing factor)-TFE3, or PRCC (papillary renal cell carcinoma)-TFE3. These proteins are thought to play an oncogenic role by activating the MET signaling pathway [5].
Because we could not detect mutations in either PKD1 or PKD2 using targeted exome sequencing, the pathogenetic mechanism of Xp11.2 translocation in relation to PKD genes could not be further examined. This may have been due to the technical difficulty of targeted exome sequencing in detecting large deletions or translocations. However, rapid progression after a long period of indolence suggested additional genetic modifying factors in our patient. An alternative genetic tool such as RNA-sequencing might reveal the pathogenesis. Our case demonstrates that Xp11.2 translocation RCC should be considered in the differential diagnosis of renal cystic masses in younger ADPKD patients without advanced CKD.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





