


INTRODUCTION
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease characterized by pathogenic autoantibodies and uncontrolled inflammatory response [1]. It often affects young women during childbearing periods; however, it can also occur at any age in children, the aged, and males [2]. Inflammatory reaction in SLE can target various organs; its symptom manifestations are skin rash including butterfly-shaped malar rash, oral ulcer, arthritis, hemolytic anemia, and nephritis. Moreover, its severity is diverse, from no detectable symptoms or no organ damage to serious and irreversible tissue damage and even death. The organs affected and disease activity are considerably different between individuals and changes as time progresses. For example, it is possible that a patient with lupus nephritis with heavy proteinuria several years ago would now show little symptoms and minimal disease activity. SLE management should be sensitive toward such changes and adjust medications accordingly.
The immunologic abnormalities in patients with SLE have been revealed. The presentation of autoantigens by antigen-presenting cells (APCs), such as macrophages and dendritic cells, to T cells leads to the maturation and release of proinflammatory cytokines. B cells produce autoantibodies forming immune complexes that deposit on tissues and damage them. Upregulation of the type I interferon (IFN) system by stimulated plasmacytoid dendritic cells enhances the maturation or function of immune cells and suppresses the differentiation of regulatory T cells maintaining self-tolerance [3,4].
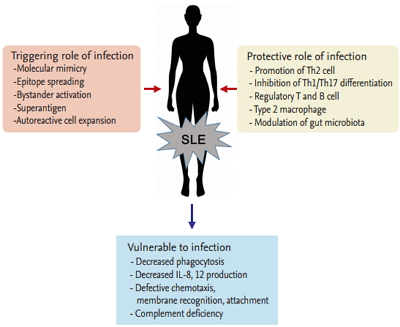
There have been some long-standing questions about infection and SLE, including whether both diseases are affected by each other, which pathogens cause SLE or are vulnerable to SLE, and how infection and lupus flare can be discriminated if patients with lupus present fevers. It is currently evident that infection affects the development of SLE, and infection occurrence and prevention are also influenced by SLE (Fig. 1). The indications for their close relationship are described here.
INFECTION AS A TRIGGERING FACTOR FOR SLE
Infectious agents, including viruses, bacteria, parasites, and fungus, are a pivotal factor for induction of autoimmunity. Many pathogens are known to contribute toward abnormal immune responses in genetically susceptible individuals through molecular mimicry, epitope spreading, bystander activation, or other mechanisms [5].
One of the reasons that autoreactive T and B cells are triggered by infection is molecular mimicry, in which there is a structural similarity between microbial peptides and self-antigens [6]. The cross-reactivity of microbial peptides and self-peptides allows the expansion of microbial-specific T cells that can respond to self-peptides [2]. Pathogens produce superantigens that bind to the variable domain of T cell receptors and various major histocompatibility complex class II molecules. Subsequently, a large number of T cells with different antigenic specificities are activated and lead to autoimmune reactions. The presentation of self-antigens by APCs cause the priming of a number of T cells by epitope spreading, overprocessing, and overpresentation of self-antigens. As T cells are activated, cytokine production increases autoreactive or memory T cell expansion via a process called bystander activation. In particular, viral infections such as hepatitis C infections target B lymphocytes and induce them to produce antibodies, leading to the formation of immune complexes. Moreover, the impaired clearance of apoptotic cells results in the insufficient removal of infectious agents [7,8]. Nuclear materials derived from apoptotic cells are exposed to the immune system, causing the production of autoantibodies or activation of autoreactive lymphocytes. Pathogens including bacteria and viruses interact with intracellular toll-like receptors (TLRs) through intracellular signaling pathways, leading to the expression of type I IFN genes. Type I IFN signatures are major pathological indicators of autoimmune diseases such as SLE, Sjogren’s syndrome, and polymyositis [9].
Viral infection
Epstein-Barr virus (EBV) or human herpesvirus 4 is a ubiquitous virus that infects about 95% of the world’s population [10]. EBV infection is known to occur during childhood without any symptoms; however, it causes infectious mononucleosis in adults and is characterized by skin rash, arthralgia, renal disorders, cytopenia, pharyngitis, and lymphadenopathy. Many studies have found that EBV infection is related to systemic autoimmunity [10]. SLE patients had higher titers of anti-EBV antibodies, and the prevalence of EBV infection in patients with SLE was higher than in the healthy population [11-13]. It was revealed that EBV showed increased replication with activation of the lytic cycle in patients with SLE, and EBV-infected B cells might become resistant to apoptosis. Moreover, EBV infection causes T cells to produce more IFNs that are pivotal proinflammatory cytokines for systemic autoimmunity. Components of the EBV protein, including Epstein-Barr nuclear antigen-1, showed similarity to autoantigens in SLE; thereby, suggesting molecular mimicry as a potential mechanism of antibody cross-reactivity and disease pathogenesis [14]. In a recent study, antibodies of early EBV infection were detected at higher levels in patients with SLE than in healthy controls, and the reactivation of EBV seemed to correlate with lymphopenia [15].
Human endogenous retroviruses (HERVs) were integrated into the human genome about 30 to 40 million years ago, and the endogenous retroviral sequence (ERS) encodes self-antigens that contribute toward immunological tolerance breakdown [16,17]. Several haplotypes of human ERS, such as human T cell lymphotropic virus 1-related endogenous sequence (HRES-1), were revealed to affect the development of SLE [18]. HERV-E 4-1 mRNA expression was higher than healthy controls and correlated with disease activity in SLE, and SLE CD4+ T cells had decreased methylation levels of the long terminal repeats in HERV-E 4-1 [19].
Parvovirus B19 infection is generally asymptomatic or self-limited; however, it can present a systemic immune response mimicking SLE flare, including arthritis and skin rash [20]. Parvovirus B19 infection induces T cell activation through molecular mimicry, leading to tissue damage and cell necrosis. Ku80, a lupus autoantigen that consists of nuclear DNA-binding proteins, functions as a specific B19 capsid-binding surface protein. The disease-related epitope binds with viral particles and activates the immune system.
Cytomegalovirus (CMV) is a member of the herpesvirus family, and it is commonly transmitted during childhood with self-limiting, or no symptoms [21]. CMV infection typically presents severe organ-specific complications in the immunosuppressed host. Although the relationship between EBV or parvovirus B19 infection and SLE seems consistent, the correlation between CMV infection and SLE development has been controversial [12,22]. Nevertheless, in recent studies, the detection of specific human CMV strains and CMV antibodies were higher in patients with SLE than in healthy controls [15,23].
Bacterial infection
Bacterial lipopolysaccharides or nucleic acid-containing immune complexes engage and deteriorate the immune system [14]. Bacterial products that are called pathogen-associated molecular patterns bind to TLRs or other receptors on APCs, B cells, and T cells. Their interactions activate immune cells to produce proinflammatory cytokines that lead to innate immunity activation. TLR ligands stimulate plasmacytoid dendritic cells to produce IFN, resulting in release of autoantibodies. In addition, tissue injuries by bacteria produce cellular debris such as nuclear components that can act as autoantigens. The release of cholera toxin B by Vibrio cholera was revealed to produce autoantibodies and promote glomerulonephritis in lupus-prone mice by enhancing lipid raft aggregation in T cells [24].
INFECTION AS A PROTECTOR AGAINST SLE
Infection by certain parasites or bacteria has been known to confer protection against autoimmune or allergic diseases; this is known as the “hygiene hypothesis” [25-27]. This hypothesis was introduced because the prevalence of allergic and autoimmune diseases showed a steep increase over the past few decades; however, background studies have not been conducted until now. The intricate mechanism involved includes promotion of T-helper 2 (Th2) and inhibition of Th1/Th17 differentiation, amplication of T-regulatory (Treg) and B-regulatory (Breg) cells and type 2 macrophages, orientation of dendritic cells toward a tolerogenic phenotype, downregulation of type 2 innate lymphoid cells, and modulation of gut microbiota. These beneficial microbes have been called ‘old friends,’ which means they exert an immunomodulatory and immunosuppressive action on the mammalian immune system [28].
Before the hygiene hypothesis was introduced, the protective role of infection against autoimmune disease development has been investigated. In lupus-prone mice, persistent infection with lactate dehydrogenase virus results in lower autoantibodies and protection from nephritis [29]. The immune response generated by malarial infection (Plasmodium falciparum) was shown to confer protection against the development of SLE because the gene related to malaria protection is an SLE-susceptible gene [30,31]. Malaria parasite infection of lupus mice controls B cell autoreactivity and confers protection against histopathological alterations and lupus severity [32,33]. Toxoplasma gondii infection had a protective role in preventing the development of autoimmune renal disorder in lupus-prone mice [34]. A glycoprotein secreted by parasitic helminths prevented nephritis in lupus mice model via reduction of antinuclear antibody (ANA) production and immune complex deposition [35]. Helicobacter pylori seronegativity was associated with the development of SLE in African-American women [36]. In some studies, the prevalence of hepatitis B virus (HBV) infection in patients with SLE was lower than that in non-SLE controls, thus revealing a protective role of HBV against SLE [37,38].
INFECTION AS A VULNERABLE POINT IN SLE
Infection is common in SLE and accounts for 25% to 50% of overall mortality [39,40]. Half of the patients with SLE experience severe infection, and more than 20% of hospitalizations occur from infections [41]. When superfluous immune reactivation damages diverse organs with constitutional symptoms, defense mechanisms against bacteria, viruses, or fungi become impaired in SLE. Decreased phagocytosis, reduced production of interleukin 8 (IL-8) and IL12 by polymorph nuclear cells, complement deficiency, and defective chemotaxis, membrane recognition, and attachment to microorganisms are known to predispose SLE patients to infection [42].
Bacterial infections are commonly reported in SLE and affect diverse organs, including the respiratory tract, urinary tract, and skin [41]. Streptococcus pneumonia is a common cause of respiratory infection. Staphylococcus aureus causes skin, soft tissue, bone, and joint infections and bacteremia. Escherichia coli is the most common cause of urinary tract infection; Klebsilla and Pseudomonas spp. also cause urinary tract infections.
Among the viral infections, that by varicella-zoster virus (VZV) is the most frequent in SLE patients, and can develop into herpes zoster [43]. The concomitant use of corticosteroids and immunosuppressive agents agents are regarded as triggering factors for VZV infection, and this infection is more frequent among patients with no or low SLE disease activity [44]. The prevalence of herpes zoster in SLE has increased, and risk of herpes zoster is associated with lymphopenia, increased use of corticosteroids, and most types of immunosuppressive therapy [45-47]. A poor cellular immunity, including decreased IFN-γ releasing cells and CD4+ T cells in response to VZV, is correlated with VZV infection and SLE [48]. CMV infection can be presented as CMV retinitis, CMV colitis, or CMV pneumonitis in patients with SLE, and may result in a systemic inflammation mimicking SLE [49]. When human papillomavirus (HPV) infection was assessed among patients with SLE, high-risk and multiple HPV infections were more frequent in SLE patients than in controls, and such a trend increased in patients with major organ involvement or immunosuppressant therapies [50,51]. In addition, the prevalence of abnormal squamous epithelial lesions was increased in patients with SLE with HPV infection [52].
The risk factors for infection in patients with SLE are disease activity, such as anti-double-stranded DNA (dsDNA) titer, low complement levels, nephritis and leucopenia, corticosteroids treatment at a prednisolone equivalent dose of 7.5 to 10 mg/day, corticosteroid pulse therapy, and high-dose regimen of cyclophosphamide [53-56]. Several studies showed that most infections, from mild to severe, can be associated with the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score that assesses the current disease activity of SLE. Cyclophosphamide is an alkylating agent used in severe manifestations of SLE such as lupus nephritis and mesenteric vasculitis, and it can strongly suppress the immune system. A higher dose of cyclophosphamide was associated with a higher prevalence of infection, and leucopenia after cyclophosphamide therapy also affects infection [57]. It is well known that corticosteroids increase susceptibility to infections, and their immunosuppressive effect is stronger when higher doses are used for a longer period [58]. However, especially in Korean patients with SLE who are not on glucocorticoids, increased SLEDAI, anemia, and active urinary sediment were associated with infection [56].
Tuberculosis (TB) caused by Mycobacterium tuberculosis is regarded as a serious infection that results in pulmonary damage in SLE, and its risk and prevalence are much higher in SLE when combined with a defective immune system and use of immunosuppressive drugs [59,60]. Use of glucocorticoids, arthritis, pleuritis, vasculitis, nephritis, organic brain syndrome, previous TB, and duration of SLE have been shown to be factors associated with TB in SLE [61,62]. Incidences of TB, extrapulmonary TB rates, and relapse rates were higher in Korean patients with SLE than in Korean patients with rheumatoid arthritis [63]. Before starting any potent immunosuppressive therapy, screening for latent TB should be performed in an endemic area like Korea. If a result is positive, treatment for latent TB would be needed. As a screening tool for latent TB, the tuberculin skin test shows a false-positive result owing to the mandatory BCG vaccination in Korea. Therefore, the interferon γ release assay (IGRA) has a critical role in screening latent TB among Korean patients with rheumatic diseases. Recently we reported that IGRA was in agreement with skin test results in patients with SLE, and SLEDAI score and glucocorticoid dose were associated with the indeterminate results of IGRA [64].
DIFFERENTIATION BETWEEN INFECTION AND LUPUS FLARE UP DURING FEVER
Fever is a typical symptom not only of infection but also of SLE flare up. When patients with SLE present fevers, physicians will need to determine the cause of the fever. Differentiation is important for further symptom management because each of the different cause of fever, infection or lupus flare up, requires directly opposite treatment from each other. If patients have an infection-related fever, they need antimicrobial agents, whereas if patients have an SLE-related fever, they need immunosuppressive therapy including steroids. Generally, infection is developed from a definite origin, and immune-related symptoms such as arthritis and skin rash are presented with fever in active SLE. If an evident source of infection exists, designing a treatment plan is easy; however, patients with fever typically show no infection source or an ambiguous disease manifestation. For a detailed and exact analysis of infection and disease manifestations, several laboratory findings are helpful to differentiate between the two conditions (Table 1). Since C-reactive protein (CRP) is unresponsive to non-infectious inflammation in SLE, an elevated level of CRP suggests a combined infection in patients with SLE [56,65,66]. In our recent study on Korean patients with lupus, CRP levels higher than 1.35 mg/dL indicated the presence of an infection with 100% sensitivity and 90% specificity [66]. Leukocyte count is elevated in SLE with infection, and remains unchanged or decreased in SLE with flare [67]. Complement levels and anti-dsDNA antibody titer are changed only in SLE with flare, but the changes are usually not considerable at already abnormal levels. Overall, CRP is the most critical marker in differentiating between infection and disease flare up in SLE. In a recent study, fever duration, anti-dsDNA antibody titers, and CRP were regarded as the most reliable markers to distinguish between infection and SLE activity; a flare risk predictive calculator using these markers was proposed [68]. In addition, neutrophil CD64 expression (or Fcγ receptor I), percentage of circulating CD27 high plasma cells, S100A8/A9 protein, and procalcitonin have been suggested and studied as new promising biomarkers to assist clinical decision-making [69,70].
INFECTION AS A TARGET OF VACCINE IN SLE
Vaccination is considered an essential method to prevent infections or reduce complications in SLE, although the clinical effects are not prominent [71]. According to the European League Against Rheumatism recommendations for vaccination in adult patients with autoimmune inflammatory diseases, vaccination status needs to be determined during the initial investigation, and it includes Haemophilus influenzae b, hepatitis A virus, HBV, HPV, influenza, Neisseria meningitidis, rubella for women of childbearing age, S. pneumonia, and tetanus toxoid [72]. Vaccinations should be administered during stable disease and before B cell depletion therapy (Table 2). However, vaccines are thought to trigger immune system activation because they stimulate an antigen-specific immune response similarly to the molecular mimicry described previously [73]. There were several reports on SLE development or flare up after vaccination, and numerous cases of SLE in relation with HBV vaccination have been reported during the 1990s. Their causality was not clearly identified, but the temporal connection of vaccination with symptom development was consistent. On the contrary, it has been observed that the weaker immunologic response presents a protective function against pathogens in patients with SLE than in healthy controls after vaccination [74,75]. The safety of vaccines in patients with SLE has been demonstrated; a recent case-control study showed that immunization is not associated with SLE development [76-78]. Therefore, vaccinations against diseases including HBV, HPV, pneumococcal, and influenza are recommended for patients with SLE in remission period. An inactive live vaccine such as zoster vaccine should be avoided when the patients are on immunosuppressive drugs such as cyclophosphamide, methotrexate, and corticosteroids (over 20 mg/day of prednisolone or equivalent).
CONCLUSIONS
Among the environmental factors causing or aggravating SLE, infection is most well described; however, how it works or who is vulnerable to it remains unclear. Currently, several viruses including EBV, parvovirus B19, and retrovirus may contribute to SLE development. In addition, certain pathogens such as T. gondii, Plasmodium, and H. pylori may play a role in preventing autoimmune inflammation.
It is evident that patients with SLE are vulnerable to infection because of the nature of the disease and symptom management practices that suppress one’s immunity. When patients with SLE present fevers, SLE-related manifestations and disease activity markers indicate an SLE flare up, and increased CRP and leukocyte count suggest a combined infection. Despite its concerns in SLE, vaccination is indispensable for patients with this disease.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





