


To the Editor,
Relapsing polychondritis (RP) is an uncommon, chronic multisystem disorder characterized by recurrent episodes of cartilaginous tissue inflammation. It can be life threatening, debilitating, and difficult to diagnose [1]. Cartilaginous tissues are the primary targets of destruction, but immune damage can spread to involve non-cartilaginous tissues such as the kidney and blood vessels [2]. Central nervous system (CNS) involvement is rare, and can manifest as cranial neuropathies, meningitis, or meningoencephalitis [3]. Only a few patients with RP and nervous system involvement have been reported in Korea. In this report, we describe a patient with multiple cranial nerve palsies due to RP related inflammatory pseudotumor.
A 60-year-old man was diagnosed with RP when he presented with auricular chondritis, hearing loss, and saddle nose deformity. He had a history of recurrent inflammatory episodes involving auricular and nasal cartilage, as well as a history of conjunctivitis. Laboratory evaluation revealed elevated C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). Autoantibody tests for rheumatoid factor, anti-nuclear antibodies, and anti-neutrophil cytoplasmic antibodies were negative. The diagnosis of RP was established based on typical clinical manifestations, without pathological examination. The patient was treated with 1 mg/kg/day of oral prednisolone and experienced symptomatic improvement. With gradual prednisolone tapering, azathioprine was added. During a 3-month follow-up, he remained well while continuing to receive 5 mg prednisolone and 100 mg azathioprine per day orally.
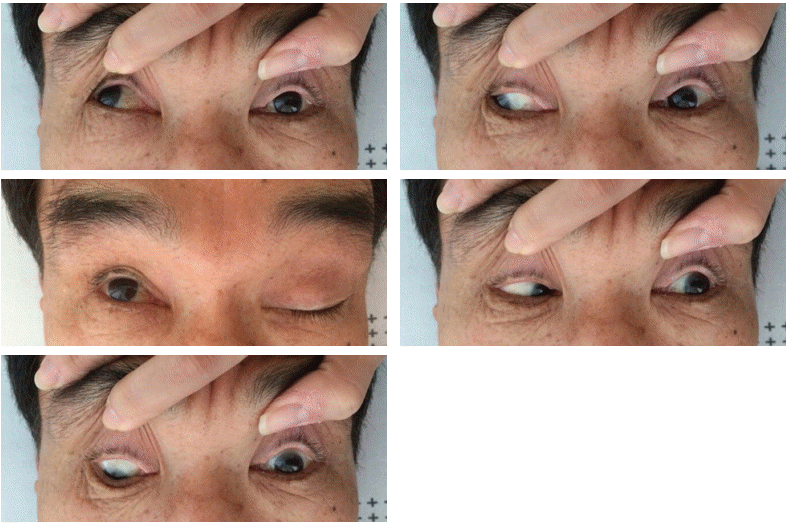
Four months after the diagnosis of RP, the patient began experiencing visual difficulties. He developed ptosis and diplopia in the left eye without any history of head trauma or ocular injury. When he was evaluated in the emergency room, his vital signs were within normal limits. Neurological examinations showed third, fourth, and sixth cranial nerve palsies of the left eye (Fig. 1). Pupils were equal in size and responded normally to light. The patient also complained of paresthesia in the territory of the first division of the left trigeminal nerve. Except for these neurologic abnormalities, his physical findings were non-specific.
Blood tests showed normal CRP and ESR at 0.08 mg/dL and 10 mm/hr, respectively. Lumbar puncture revealed an opening pressure of 14 cm H2O. The cerebrospinal fluid (CSF) tests results were white blood cell count, 0/mm3; glucose, 70 mg/dL (plasma glucose level 100 mg/dL); protein, 23.7 mg/dL, adenosine deaminase 0.1 IU/L. No organisms were observed on Gram stain and acid-fast bacillus stain. Cryptococcus antigen test and mycobacterium tuberculosis polymerase chain reaction test were negative. The final CSF culture results revealed no organisms. CSF cytology was negative for malignant cells.
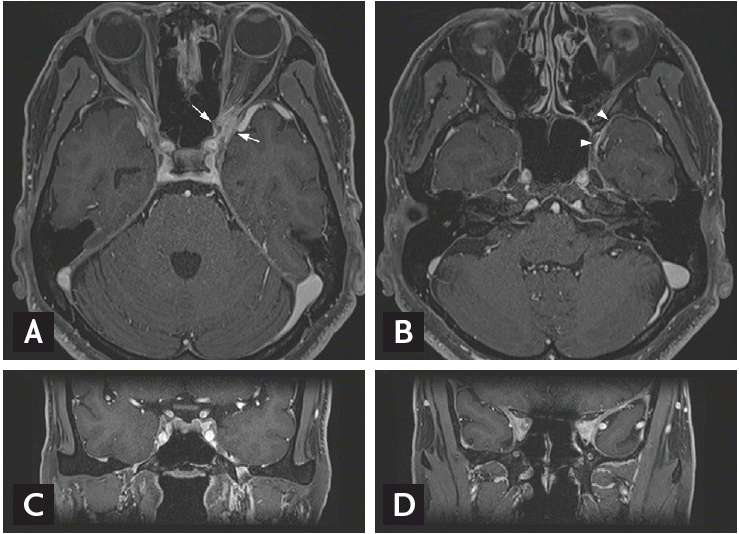
Magnetic resonance imaging (MRI) of the cranial nerves showed diffuse thickening and enhancement of the bilateral cavernous sinuses, orbital apex, and pachymeninges along with MeckelŌĆÖs cave (Fig. 2). These findings suggest inflammatory pseudotumor involvement of the cranial nerve pathways. Nerve conduction study, electromyography, and blink test were performed. The results indicated bilateral trigeminal nerve dysfunction. Biopsy of the lesion was not performed because it was difficult to reach and the procedure was considered high risk for complications such as nerve injury.
The patient was started on intravenous (IV) methylprednisolone, 1 mg/kg/day. After 4 days, extraocular movements were slightly improved, but the left eye ptosis remained. He was started on IV methylprednisolone, 1 g/day over 5 days followed by oral prednisolone, 60 mg/day. High dose steroid therapy resulted in full neurologic recovery after 3 weeks. The prednisolone dosage was gradually reduced.
Few previous cases of RP with CNS involvement have been reported. In most cases, the clinical manifestation was meningoencephalitis. Other associated neurological disorders include impaired cognitive dysfunction, seizure, ptosis, and diplopia. The etiology of CNS involvement in RP patients remains unknown but likely originates from autoimmunity. Wang et al. [3] speculated that vasculitis or non-specific inflammation might involve the leptomeninges and brain parenchyma.
Ocular manifestations of RP are diverse. The most common manifestations are scleritis and episcleritis. Iritis may occur in as many as 30% of patients who present with scleritis or keratitis. Proptosis with chemosis simulating an orbital pseudotumor is the uncommon manifestation, but may be the initial presentation of RP [4]. Extraocular muscle palsies have been reported and are probably related to a vasculitis involving muscles or related to nerve insults.
The current patient presented with multiple cranial nerve palsies and without any findings of meningoencephalitis or scleritis. MRI revealed diffuse thickening and enhancement of the cranial nerve pathways including bilateral cavernous sinuses, orbital apex, and MeckelŌĆÖs cave.
If an orbital mass is seen, biopsy may be necessary to reveal the type of inflammatory histology and exclude neoplastic causes. In this case, a biopsy was not performed for tissue confirmation because of the lesion location. Lichauco et al. [5] reported an orbital mass in a patient with RP that was confirmed by biopsy as mucosa-associated lymphoid tissue type B cell lymphoma. In the reported case by Lichauco et al. [5], biopsy was performed because the initial steroid treatment was only partially effective. Our patient completely recovered after steroid treatment, but biopsy would be necessary if his symptoms reappear or if any suspicion of malignancy arises.
We report our experience with a patient diagnosed with RP and pseudotumor who presented with multiple cranial nerve palsies. In RP patients, CNS and ocular manifestations are diverse. If neurologic or ophthalmologic symptoms develop, careful neurologic examination, imaging, and biopsy should be considered to establish a diagnosis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





