


INTRODUCTION
The 5'-adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a serine/threonine protein kinase that acts as a cellular energy sensor to regulate cellular energy homeostasis by monitoring intracellular AMP/adenosine triphosphate (ATP) ratios. Activating AMPK inhibits the proliferation and growth of various cancer cells [1,2,3]. Some studies have demonstrated that the antitumor properties of 5-aminoimidazole-4-carboxamide-ribonucleotide (AICAR), a strong AMPK activator, are mediated by genetic mutations or are regulated in a cell-type specific manner [4,5,6].
AMPK contributes to regulation of the mitogen-activated protein kinase (MAPK) pathway [7,8,9,10,11]. Given that these kinases are tightly related to cellular responses, including cell proliferation, differentiation, growth, and apoptosis, the cross-talk between MAPK and AMPK has been investigated as a key mechanism by which cancer progression is regulated. Studies have demonstrated that activating AMPK by AICAR is associated with regulation of the MAPK pathway [12,13]. We reported the relevance of AICAR-mediated AMPK activation according to BRAF mutation status in thyroid cancer cells. We suggested that thyroid cancer cells harboring the BRAFV600E mutation are more susceptible to the anticancer effects of AICAR than those with wild-type BRAF [6]. However, the extent of the cross-talk between AMPK and MAPK in human thyroid cancer cells remains to be established.
Members of the MAPK family include extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 MAPK. The ERK pathway has been implicated in regulating cellular responses, such as proliferation and differentiation, as well as protection against apoptosis. In contrast, the JNK and p38 pathways are key mediators of stress-dependent apoptosis and inflammatory responses [14,15]. In this study, we evaluated the possible involvement of MAPK in the activation of AMPK by AICAR in FRO thyroid cancer cells.
METHODS
Reagents and cell culture
AICAR was purchased from Toronto Research Chemicals (North York, ON, Canada). SB203580, a specific p38 MAPK inhibitor, was purchased from Calbiochem (San Diego, CA, USA). Activated and pan-type antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), and a monoclonal antibody directed against p21 was obtained from BD Pharmingen (San Jose, CA, USA). Horseradish peroxidase-conjugated antimouse and rabbit antibodies were purchased from Vector Laboratories (Burlingame, CA, USA). All other biotechnology grade chemicals were purchased from Amresco Inc. (Solon, OH, USA).
FRO human thyroid cancer cells were cultured in RPMI 1640 containing 10% fetal bovine serum at 37Ōäā in 5% CO2. The cells were seeded at 1 ├Ś 103 cells/well in 96-well culture plates or at 5 ├Ś 105 cells/60-mm culture dish, and incubated for 24 hours before treatment with AICAR. In some experiments, the cells were pretreated with 20 mM SB203580, a specific p38 MAPK inhibitor, for 30 minutes before adding AICAR.
Cell viability assay
Cell viability was measured using the colorimetric Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) assay in accordance with the manufacturer's instructions. Cells cultured in 96-well culture plates were treated with various concentrations of AICAR, with or without SB203580, for 48 or 72 hours. The amount of formazan dye generated by cellular dehydrogenase activity, an indicator of cell viability, was determined by measuring absorbance at 450 nm using a SPECTRA MAX Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Untreated cells served as controls.
Western blotting
Aliquots (30 mg) of total cellular protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 10% to 12% (w/v) gradient NuPAGE gels (Invitrogen, Carlsbad, CA, USA), transferred to nitrocellulose membranes (Amersham Bioscience, Piscataway, NJ, USA), and incubated with specific antibodies. Immunoreactive proteins were detected by enhanced chemiluminescence (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and relative protein band intensity was determined by densitometric scanning using a FluorS Multi-Imager (Bio-Rad, Hercules, CA, USA). Expression levels of target proteins were normalized to that of ╬▓-actin in each sample.
Design of siRNA targeting AMPK-╬▒1 and transfection
The small interfering RNA (siRNA) sequence targeting the AMPK-╬▒1 gene (GenBank accession no. NM_206907) was designed using the BLOCK-iT RNAi Designer software (Invitrogen). Three potential AMPK-╬▒1-targeting siRNAs (Invitrogen Stealth RNAi siRNA) were used at the same concentration to screen for effective siRNAs. The sequences of selected siRNAs were 5'-UUA AGG CUU CAU CAU CAA UCA UGG U-3' and 5'-ACC AUG AUU GAU GAU GAA GCC UUA A-3'. Nonsilencing random sequence siRNAs were purchased from Invitrogen and used as negative controls. A random oligomer conjugated to green fluorescence protein (Invitrogen) was used to assess transfection efficiency.
Transfections were performed using the Lipofectamine RNAi MAX reagent (Invitrogen), according to the manufacturer's instructions. An efficiency test and negative control transfections were performed using the same reagents. After transfection, the cells were incubated for 24 hours and treated with AICAR in the presence or absence of SB203580. Changes in expression of AMPK-╬▒1 or other molecules were evaluated using Western blot analysis.
RESULTS
AICAR inhibits growth and induces death in thyroid cancer cells
AICAR is the best characterized AMPK activator, and promotes apoptosis in various cancer cells. AICAR acts in a cell-type-specific manner, and AMPK-independent effects of AICAR have been reported. We first investigated the effect of AICAR on cell growth and whether this correlated with AMPK activation in FRO thyroid cancer cells.
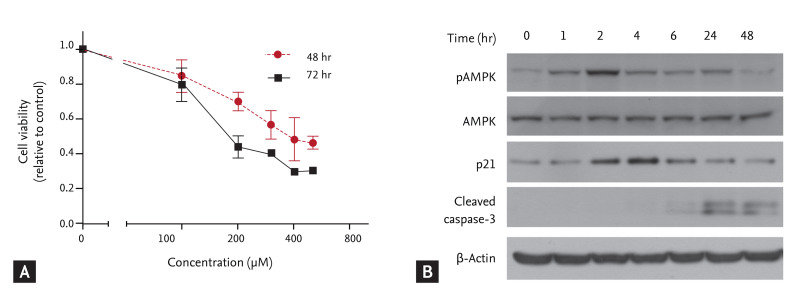
The cells were incubated with different concentrations of AICAR for 48 or 72 hours, and cell proliferation was determined using a colorimetric CCK-8 assay. AICAR significantly inhibited proliferation of FRO cells in a time- and dose-dependent manner (Fig. 1A). We used 200 ┬ĄM AICAR, which corresponded roughly to the IC50 in a FRO cell viability assay, and performed a Western blot analysis to determine whether AICAR stimulated activation of AMPK and proapoptotic molecules. As shown in Fig. 1B, AICAR treatment promoted rapid AMPK activation and p21 accumulation, and also enhanced cleavage of caspase-3. These results suggest that AICAR not only triggers AMPK activation but also suppresses cell growth by inducing p21 and activating caspase-3.
AICAR inhibits cell growth by activating p38 MAPK in thyroid cancer cells
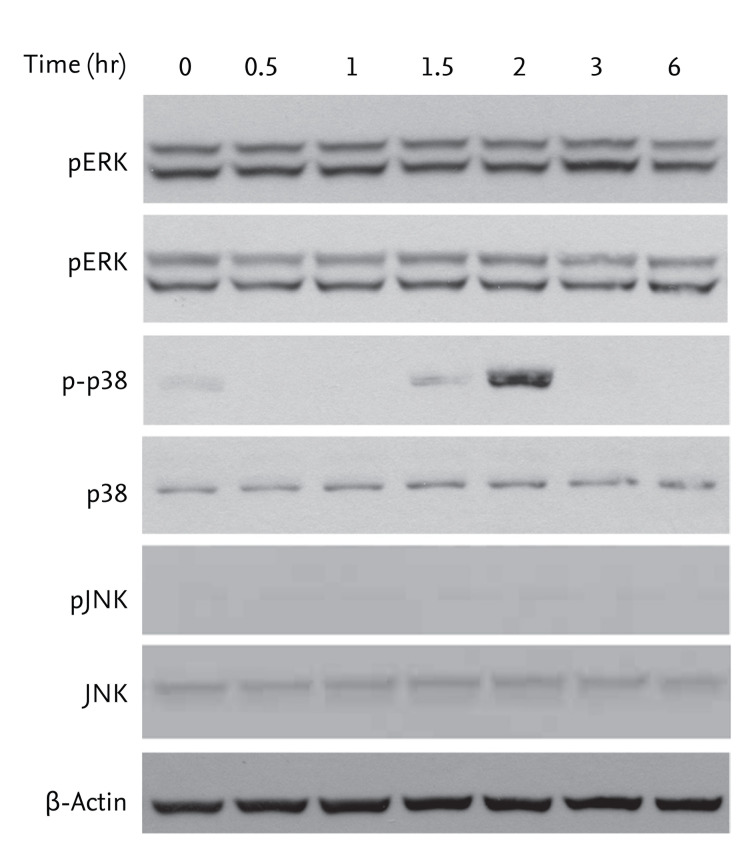
To establish whether activation of AMPK by AICAR is related to MAPK activity, cells were treated with 200 ┬ĄM AICAR for the indicated times. We evaluated changes in the phosphorylation status of MAPK molecules (ERK, p38, and JNK) by Western blot analysis. AICAR treatment significantly promoted activation of p38 MAPK, but not ERK or JNK in FRO cells (Fig. 2).
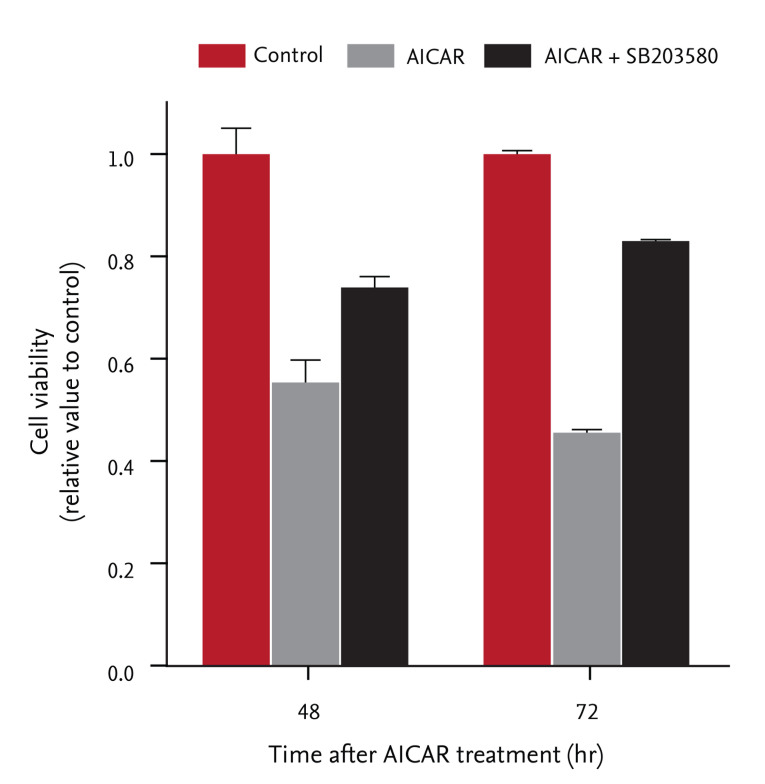
We examined whether inhibition of cell proliferation by AICAR was associated with activation of p38 MAPK in FRO cells. Cells were pretreated with SB203580 (20 ┬ĄM), a specific p38 MAPK inhibitor, for 30 minutes, and then incubated with 200 ┬ĄM AICAR for the indicated times. After incubation, cell proliferation was assessed using the colorimetric CCK-8 assay. As shown in Fig. 3, AICAR treatment suppressed relative cell proliferation in a time-dependent manner, but pretreatment with SB203580 significantly rescued cells from AICAR-induced cell death (19% increase at 48 hours and 37% increase at 72 hours, respectively). These results indicate that activating p38 MAPK is required for AICAR-induced AMPK activation and inhibition of cell proliferation in FRO thyroid cancer cells.
AMPK-╬▒1 regulates AICAR-induced cell death by activating p38 and caspase-3 in thyroid cancer cells
Given our finding that AICAR-induced AMPK activation also induced cell death by activating p38 MAPK in FRO cells, we elucidated the involvement of AMPK in AICAR-induced activation of p38 and caspase-3. We performed RNA interference experiments using siRNAs targeting AMPK in FRO cells. AMPK is a heterotrimeric complex that consists of three subunits, including catalytic serine/threonine kinase subunits (contains ╬▒1 and ╬▒2) and two regulatory subunits (╬▓ and ╬│) [16]. We first investigated the expression patterns of the a1 and a2 catalytic subunits of AMPK in FRO cells. As shown in Fig. 4A, AMPK-╬▒1 was predominantly expressed, whereas AMPK-╬▒2 was barely expressed in these cells. As a consequence, we next assessed the efficiency of siRNA-mediated silencing of AMPK-╬▒1. Cells were transfected with AMPK-╬▒1-targeting siRNAs or with random RNAi oligomers (negative control), and then analyzed for AMPK expression (Fig. 4B). Transfection of AMPK-╬▒1-targeting siRNAs markedly reduced the level of AMPK protein compared with mock treatment (no oligomer) and random oligomers for up to 96 hours after transfection. We further examined the effects of AMPK-╬▒1 on AICAR-induced p38 activation and cell death regulatory molecules in FRO cells. The cells were transfected with AMPK-╬▒1-siRNAs for 24 hours, followed by stimulation with 200 ┬ĄM AICAR for the indicated times. As expected, AMPK-╬▒1-siRNAs inhibited AICAR-induced p38 activation, and reduced protein abundance of cell death regulators such as p21 and caspase-3 (Fig. 4C). Taken together, these findings indicate that AMPK-╬▒1 is directly involved in AICAR-induced p38 activation and in the initiation of cell death by activating p21 and caspase-3 in FRO thyroid cancer cells.
DISCUSSION
We demonstrated previously the relevance of AMPK activation with regard to AICAR and BRAF mutation status in thyroid cancer cells. In this study, we suggest that thyroid cancer cells harboring a BRAF mutation are more susceptible to the effects of AICAR than cells harboring wild-type BRAF [6]. We investigated whether activating AMPK with AICAR was related to other aspects of MAPK signaling in thyroid cancer cells that harbor the BRAFV600E mutation. Specifically, we investigated whether p38 MAPK acts downstream of the AICAR-stimulated AMPK pathway in FRO thyroid cancer cells that harbor the BRAFV600E mutation. We first showed that AICAR promotes AMPK activation and cell death by inducing accumulation of p21 and activating caspase-3. We next examined whether cell death caused by AICAR was associated with MAPK activation. Interestingly, AICAR treatment significantly activated p38 MAPK, but not ERK or JNK MAPK, in FRO cells. Furthermore, SB203580, a specific p38 inhibitor, partially but significantly reduced the rates of AICAR-induced cell death in FRO cells. These results indicate that p38 MAPK activity is involved in the regulation of AICAR-induced AMPK activation and cell death in FRO cells. However, we did not evaluate the effect of AICAR in other thyroid cancer cells. Whether p38, p21, and caspase-3 also participate in activating AMPK by AICAR in BRAF wild-type thyroid cancer cells awaits future studies.
Both the AMPK and the MAPK pathways act as key regulators of cell growth, proliferation, and differentiation during cancer progression. Many studies have investigated the signaling network involving AMPK and MAPKs in human cancers. In one study, the berberine-induced activation of AMPK reduced ERK activity and thereby inhibited the metastatic potential of human melanoma cells [10]. The AMPK activator metformin induces apoptosis in lung cancer cells by activating both the JNK and p38 MAPK pathways [17]. Activation of AMPK by immunotoxin induces reactive oxygen species accumulation and activates the stress-activating kinases JNK and p38 MAPK in MA11 breast cancer cells [8]. Several studies have used AICAR as an effective AMPK activator to elucidate the signaling network that links AMPK to MAPKs in various types of cancer [7,9,18,19].
AMPK is activated by upstream kinases, such as liver kinase B1 and calcium/calmodulin-dependent protein kinase under various stress conditions, including oxidative stress, hypoxia, and ischemia [20,21]. Activation of AMPK by phosphorylation of threonine 172 in its catalytic subunit enables it to regulate metabolic processes, cell proliferation, and cell cycle progression [21]. In addition, the ability of AMPK to inhibit the mammalian target of rapamycin or AKT signaling pathways suggests that it is a key regulator of cell proliferation and survival in diverse malignant cell types [22,23,24]. Several studies have shown that AMPK activity is correlated with the metabolic status of various cancers [16,25,26]. The reduced activation of AMPK and its substrate acetyl CoA carboxylase is correlated with higher histological grade and node metastasis of breast cancer [26].
We performed RNA interference experiments using siRNAs targeting AMPK-╬▒1 in FRO cells, instead of compound C, which has been used as a selective inhibitor of AMPK [27,28]. We tested the effects of compound C on AICAR-induced AMPK activation and cell death in FRO thyroid cancer cells. Although compound C clearly inhibited AICAR-induced AMPK activation, it had no effect on p38 activation or cell-death-induced AICAR treatment. Compound C inhibits not only AMPK activation but also a number of other kinases independently of AMPK inhibition [29]. Compound C triggers cell death in MCF7 breast carcinoma cells independently of the AMPK pathway [30]. In this context, although our present results show that compound C inhibited AMPK in FRO cells, it is possible that inhibition of AMPK by compound C involves cell growth inhibition through other mechanisms.
Given that the levels of AMPK-╬▒1 exceed those of AMPK-╬▒ in FRO cells, we used siRNAs directed against AMPK-╬▒1 in our RNA interference experiments in these cells. AMPK-╬▒1-targeting siRNAs were efficiently transfected into cells and significantly inhibited the expression of this gene for up to 96 hours after transfection. Our observations indicate that AMPK-╬▒1 RNA interference inhibited AICAR-induced activation of p38 and the accumulation of cell death regulatory molecules such as p21 and caspase-3.
In conclusion, our data demonstrate that AICAR inhibits cell proliferation and survival by activating AMPK in FRO cells. Activation of p38 and proapoptotic molecules following AMPK activation is also involved in the effect of AICAR. This result suggests that the p38 MAPK pathway may be a downstream target of the AICAR-mediated activation of AMPK in thyroid cancer cells. Although the details of the signaling network that connects AMPK and MAPKs remain largely unknown, our findings regarding the regulation of AMPK activity provide important new insights into thyroid cancer therapies using AMPK activators.
KEY MESSAGE
1. Activating 5'-adenosine monophosphate (AMP)-activated protein kinase (AMPK) with 5-aminoimidazole-4-carboxamide-ribonucleotide (AICAR) inhibits thyroid cancer cell proliferation and induces apoptosis.
2. AICAR induces AMPK-╬▒1-mediated activation of p38 mitogen-activated protein kinase (MAPK) and accumulation of p21 in thyroid cancer cells.
3. AMPK activators could be a potential therapeutic target for thyroid cancer.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





