


INTRODUCTION
The etiology of delayed puberty is heterogeneous and includes: constitutional delay, hypogonadotropic states, hypergonadotropic states as well as chronic illness1). Hypopituitarism is not a common cause of delayed puberty2). The current approach to the diagnosis of hypothalamic hypophyseal lesions using a combined pituitary function stimulation test and pituitary MRI have improved detection and allowed for the diagnosis of hypopituitarism as a cause of delayed puberty2).
We report a case of a 22-year old man who presented with absent secondary sexual characteristics and was otherwise asymptomatic. Using a combined approach with the pituitary function stimulation test and pituitary MRI, we could identify the cause of delayed puberty to be hypopituitarism due to a pituitary structural abnormality, i.e., pituitary stalk dysgenesis and ectopic neurohypophysis. The patient was started on hormone replacement therapy and gradually developed secondary sexual characteristics.
CASE REPORT
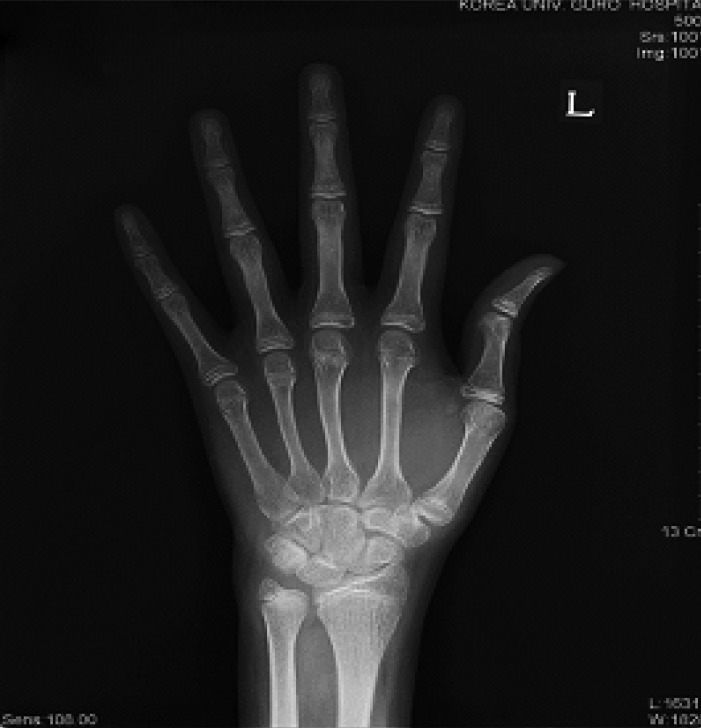
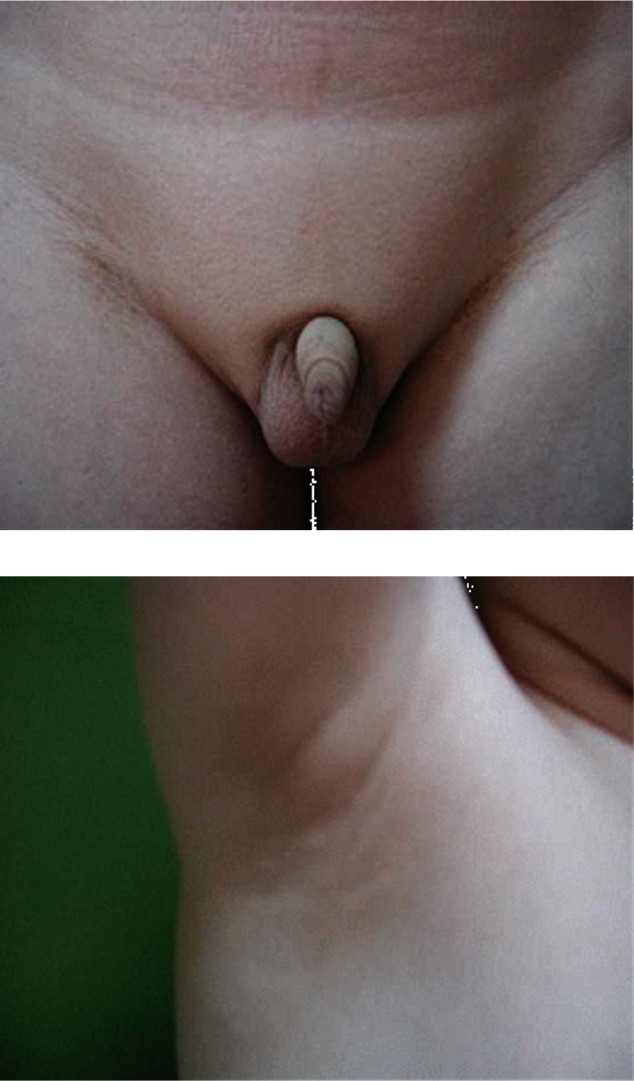
A 22-year old man was referred to our clinic because of the absence of the development of secondary sexual characteristics. The past medical history was unremarkable there were no significant pregnancy or perinatal problems. The family history was negative for growth failure, pituitary or thyroid disease. During childhood, the patient reported that he had always been short for his age. However, after a delayed growth spurt, he began to grow steadily at 20 years of age. At the time of admission, he was 183 cm. He weighed 75 kg and his body mass index (BMI) was 22.4 kg/m2. He had normal intelligence and normal body proportions. His bone age was 15 years, which was slightly below expected. X-ray evaluation of both hands showed open epiphysis (Figure 1). Physical examination revealed a micropenis, no pubic or axillary hair and palpable though small testes (Figure 2). These findings were compatible with a Tanner stage I development. The patient had a high pitched voice and appeared young for his stated age. The patient denied physical discomfort, had no signs of gynecomastia, no signs of orthostatic hypotension, nor any symptoms that could be associated with hypothyroidism.
The laboratory findings showed normal erythrocyte sediment rate, hematological parameters, blood glucose, serum sodium, potassium, calcium, phosphate, magnesium, and creatinine. Random testosterone (0.08 ng/mL, reference range 2.4~18.3 ng/mL) and luteinizing hormone (LH) (below 0.1 mLU/mL, reference range 0.4~5.7 mLU/mL) levels were all low; the follicle stimulating hormone (FSH) levels were within normal limits. The karyotype was normal, 46,XY. As a result of the random sexual hormone levels and karyotyping analysis, we could classify the delayed puberty as hypogonadotropic hypogonadism. Therefore, we performed a combined pituitary function stimulation test and pituitary MRI for further assessment. We injected regular insulin (0.1 u/Kg), TRH (200 ug), and LHRH (100 ug) and observed that 2 hours later the blood sugar fell to 60 with patient complaint of hypoglycemic symptoms. The combined pituitary function stimulation test showed no increase in serum LH, FSH, growth hormone (GH), adrenocorticotropin hormone (ACTH) level, and the serum levels of prolactin and thyroid stimulating hormone (TSH) showed a normal increment (Table 1). The MRI showed decreased pituitary stalk enhancement and an enhancing structure in the hypothalamic area, which appeared to be ectopic neurohypophysis (Figure 3A, 3B).
Therefore, considering the results of the above evaluation, the delayed puberty, in this 22 year old man, was the result of hypopituitarism due to pituitary stalk dysgenesis and ectopic neurohypophysis.
The patient was started on hormone replacement therapy with prednisolone 5 mg/day, levothyroxine 0.1 mg/day and testosterone 250 mg IM every 3 weeks. After 3 months of treatment, secondary sexual characteristics began to develop; pubic and axillary hair was noted and the voice changed to a lower pitch. The serum testosterone level, previously 0.08 ng/mL, increased to 0.57 ng/mL.
DISCUSSION
Delayed puberty is generally defined when puberty development is two standard deviations (SD) below the mean for a given population1, 3). Hypopituitarism is not a common cause of delayed puberty, but recently the incidence has increased due to improvement in diagnostic tools2). The clinical manifestations associated with hypopituitarism vary, depending on the severity of the pituitary hormone deficiency4, 5). Presentation of symptoms are variable and include: acute adrenal insufficiency and profound hypothyroidism, symptoms indicating a pituitary mass lesion; or the symptoms may be nonspecific such as: fatigue and delayed puberty, as reported in our case2, 4, 5). Therefore, hypopituitarism should be considered in all patients with abnormal development of secondary sexual characteristics even if they are otherwise asymptomatic.
In our case, the cause of delayed puberty was hypopituitarism due to pituitary dysgenesis. The pituitary gland develops as a result of a fusion of the adenohypophysis and neurohypophysis during the embryonic period. Incomplete downward migration of the neurohypophysis as a result of a genetic defect or a pituitary stalk injury, from perinatal trauma, may lead to fusion defects. Fusion defects are associated with anterior pituitary gland atrophy due to the lack of stimulation from the hypothalamus6). Craft et al. reported a high frequency of perinatal insults, such as breech delivery or neonatal hypoxemia, in children with idiopathic hypopituitarism9, 10). Fujisawa et al and Kikuchi et al. reported the findings of stalk transection and ectopic posterior pituitary at MRI in patients with hypopituitarism who had a history of perinatal insult; these findings suggested that the traumatic ischemic injury of the pituitary stalk or median eminence was the primary cause of the pituitary hormonal deficiencies7, 8). However, other studies propose a congenital cause for this form of pituitary abnormality. In these reports mutations in genes responsible for normal posterior pituitary lobe descent and stalk development are suggested to explain cases of hypopituitarism due to a pituitary structural abnormality11-15). To date, although there are possible candidates such as HESX116) and LHX4 genes17), the specific genetic abnormalities leading to pituitary stalk dysgenesis and posterior pituitary ectopia have not been identified. Future analysis is planned to examine other transcription factors and signaling molecules, important during pituitary embryogenesis, especially those involved in posterior pituitary ectopia and pituitary stalk dysgenesis.
In a high proportion of patients with non-familial idiopathic growth hormone deficiency (both isolated growth hormone defect and multiple pituitary hormone defect), characteristic radiographic findings include a) small to absent anterior pituitary gland b) small or absent pituitary stalk and c) ectopic posterior pituitary hyperintensity located at the base of the hypothalamus14).
If an enhancing structure in the hypothalamic area is identified, the diagnosis must distinguish an ectopic neurohypophysis from granulomatous disease and metastatic disease. The findings in our case suggested an ectopic neurohypophysis because of an enhancing structure that showed high signal intensity in both precontrast and postcontrast images. The presence of the neurosecretory granule containing arginine vasopressin neurophysin complex in the astrocytic glial cell can explain the high signal intensity of posterior pituitary lobe18).
In previously reported cases, many patients with an ectopic neurohypophysis had no posterior pituitary gland dysfunction. In 1996, Adamsbaum reported, in three thousand normal individuals, that the posterior pituitary gland was always located in a fixed site; however, in patients with growth hormone deficiency ectopic neurohypophysis was identified in 40~60%, and 60~80% of them had partial pituitary hormone deficiency but rarely diabetic insipidus19). This is because of the proliferation of axons and reorganization of the posterior pituitary lobe at the site above the cutting point of the pituitary stalk7). Our patient had no sign of diabetes insipidus and therefore we did not test for it with water deprivation.
Our patient had no history of abnormal gestation, breech presentation, and ischemic insult at birth. So we could assume that the structural pituitary abnormality of this 22-year old man might come from the result of congenital and genomic abnormality. However, we did no further evaluation at genomic level. The combined pituitary function testing was noted for no response for cortisol and growth hormone. However, the patient had no complaints suggestive of adrenal insufficiency and the height was within normal limits (183 cm). Extensive destruction of the pituitary gland, greater than 60~70 percent, is required for symptoms of pituitary insufficiency; when present these symptoms are diverse. As in our patient there may be no specific symptoms of steroid deficiency. However, with greater stress symptoms may become more apparent; this might occur as a result of infection or in association with a surgical procedure. Prior reports have shown that in spite of GH deficiency, due to pituitary stalk dysgenesis, height is within normal limits. D.T den Ouden et al explained such phenomenon by offering the following three hypotheses: First, because of the total absence of estradiol, the epiphyses do not close, and the patient continues to grow possibly due to other factors such as insulin. Insulin can act as a growth stimulus partially through activating the IGF-1 receptor. Second, the fact that estrogens have a slightly antagonistic effect on the bioactivity of GH could explain why, in the absence of estrogens, low GH secretion has a greater effect than expected. Last, a possible growth stimulus in patients with high prolactin (PRL) levels, in patients with low GH, which has been implicated in the pathophysiology of growth without GH in craniopharyngioma20).
In conclusion, there are many cases of partial hypopituitarism that go undiagnosed because they are asymptomatic, and only have absence of secondary sexual development. Therefore, it is important to consider the possibility of hypopituitarism, even in patients who present without other symptoms, as in our case. Furthermore, if hypopituitarism results from a structural pituitary abnormality, especially without a history of perinatal birth injury such as breech presentation, further evaluation is indicated at the molecular level to determine whether a genetic abnormality may explain the structural pituitary malformation, such as posterior pituitary ectopia and pituitary stalk dysgenesis.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





