


INTRODUCTION
Oxygen free radicals(superoxide radical, hydrogen peroxide, and hydroxyl radicals) have been considered to be responsible for the pathogenesis of ischemia-reperfusion injury and toxic chemical injury in a variety of organs including myocardium, brain, intestione and kidneys(McCord1), 1985; Bonventre et al.2), 1988; Paller et al.3), 1991). They are produced by renal cells and also by the inflammatory bone marrow-derived cells invading the renal tissue(Shah et al4), 1983; Baud and Radaillou5), 1986). Biological membranes have a high content of polyunsaturated fatty acids, which are particularly susceptible to peroxidatiove attack by reactive oxygen free radicals, resulting in lipid peroxidation (Chance et al.6), 1979; Mead7), 1976). Oxygen free radicals and lipid peroxidation can affect membrane structure(Chance et al.6), 1979; Arstila et al.8), 1972), permeability(Siflinger-Birnboim et al.9), 1992) and the function of essential proteins such as Na-K-ATPase(Kako et al.10), 1988).
In vivo studies have demonstrated a critical ole of oxygen free radicals in pathogenesis of acute glomerular injury and proteinuria during the early phase of nephrotoxic nephritis(Rehan et al.11), 1984). In in vitro models using a suspension of rat proximal tubule segments, t-butylhydroperoxide(t-BHP), a potent oxidant, induces the severity of tubular dysfunction as reflected by decreases in tubular respiration which is associated with a progressive increase in lipid peroxidation(Borkan and Schwartz12), 1989; Schnellmana13), 1988). Rush et al.14) (1985) reported, however, that lipid peroxidation did not play a role in the acute cytotoxicity of t-BHP in suspension of isolated rat hepatocytes. Thus, the precise mechanism of t-BHP-indcued cell injury remains ot be determined. This study was carrided out to determine the effect of oxygen free radicals on organic anion transport in renal proximal tubule, alterations in transport of p-aminohippurate(PAH), an organic anion, and tetraethylammonium (TEA), an organic cation, were examined in renal cortical slices subjected to t-BHP, a model hydroperoxide. The magnitude of lipid peroxidation also was assessed by measuring the endproduct, malondialdehyde (MDA).
METHODS
1. Slice Preparation
New Zealand white rabbits weighing approximately 2kg were sacrificed and the kidneys were rapidly removed. The kidneys were immediately perfused through the renal artery with an ice-cold isotonic saline solution containing 140mM NaCl, 10mM KCl and 1.5mM CaCl2, to remove as much blood as possible. Thin(0.4ŌĆō0.5mm thick) slices of renal cortex were prepared using a Stadie-Riggs microtome and were stored in an ice-cold modified Cross-Taggart medium containing 130mM NaCl, 10mM KCl, 1.5mM CaCl2, 5mM Na acetate and 20mM Tris/HCl(ph 7.8).
2. Accumulation of Organic Ions
Approximately 50mg(wet wt.) of slices were transferred into a 20ml beaker containing 4ml of the modified Cross-Taggart medium, and incubated with 75╬╝M 14C-PAH or 10╬╝M 14C-TEA (Amersham, Alrington heights, IL). The incubation was carried out for 60min in a Dubnoff metabolic shaker at 25┬░C under a 100% oxygen atmosphere.
Immediately after incubation, the slices were quickly removed from the beaker, blotted, weighed and solubilixed in 1 N NaOH. Aliquots of the incubation medium and the solubilixed tissue were pipetted into a scintillation vial contalining Aquasol(New England Nuclear) and the radioactivity was determined using a liquid scintillation counter(Packard Tricarb 300C). PAH uptake by renal slices was expressed as the slice to medium(S/M) ratio: the concentration of the compound in the tissue(mole/g wet tissue) divided by that in the medium(mole/ml medium).
3. Measurement of PAH Efflux
The efflux of PAH from cortical slices was determined as described previously by Kim et al.15) (1986). The slices were preincubated for 60 min in a medium containing [14C]PAH(75╬╝M), after which slices were rinsed for 20sec in PAH-free medium in order to remove PAH adhering to the tissue surface, thje slices were then transferred at 2-min intervals through a series of 30 beakers containing PAH-free modified Cross-Taggart medium at 25┬░C. This procedure was performed in a Dybnoff metabolic shaker with a hinged plexiglass cover and 100% oxygen atmosphere. The quantity of compoung collected from each runout chamber after exposure of the tissue and the amount of compoud that remained in the tissue after the experiment were used to construct the efflux curve and calculate the rate constants.
4. Oxygen Consumption Measurement
The oxygen consumption of renal cortical slices was measured with an oxygen monitor(Yellow Springs Instrument Co., model 53). Approximately 50mg of slices were incubated in a reaction vessel containing 4ml of the modified Cross-Taggart medium saturated with oxygen at 25┬░C. Decrease in PO2 in the medium was measured using a Clark electrode for 15min, and the rate of oxygen consumption was calculated.
5. Lactate Dehydrogenase(LDH) Measurement
Following incybation, slices were removed and homogenized in 2ml of distilled water. The tissue homogenate was centrifuged at 1,000rpm for 5min. The pellet was discarded and the supernatant was usee. LDH activity in the supernatant and incubation medium was determined using LDH measurement kit(latron Lab., Japan).
6. Lipid Peroxidation
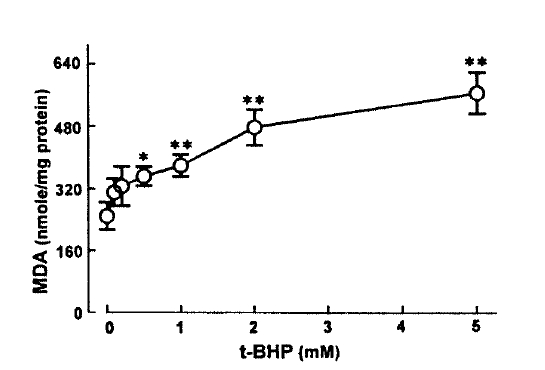
Lipid peroxidation was estimated by measuring the renal cortical content of the lipid peroxidation product, malondialdehyde(MDA) according to the method of Uchiyama and Mihara15)(1978). Following incubation, the slices were rapidly removed and homogenized in ice-cold 1.15% KCl(5% wt/vol). A 0.5ml of homogenate was added to 3ml of 1% phosphoric acid and 1ml of 0.6% thiobarbituric acid. The mixture was heated for 45 min on a boiling water bath. After addition of 4ml of n-butanol, the contents were vigorously vortexed and centrifuged at 2,000g for 20 min. The absorbance of the upper, organic layer was measured at 535 and 520nm with diode array spectrophotometer(Hewelett Packard, 8452A), and was compared to results obtained using freshly prepared malondialdehyde tetraethylacetal standards(Sigma Chemical Co.). MDA values were expressed nanomoles per mg protein. Protein was measured by the method of Bradford17) (1971)
RESULTS
1. Effect of t-BHP on Organic Ion Uptake
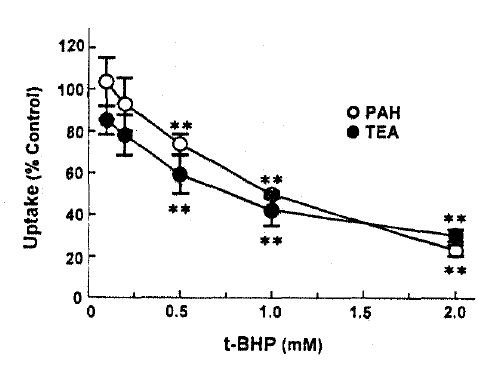
PAH and TEA uptake in cortical slices was measured in the presence of various concentrations of t-BHP in the incubation medium. The results are depicted in Fig. 1. t-BHP inhibited PAH and TEA uptake in a dose-dependent manner over the concentrations of 0.5ŌĆō2.0mM, showing 50% inhibition at 1.0 and 0.85mM, respectively.
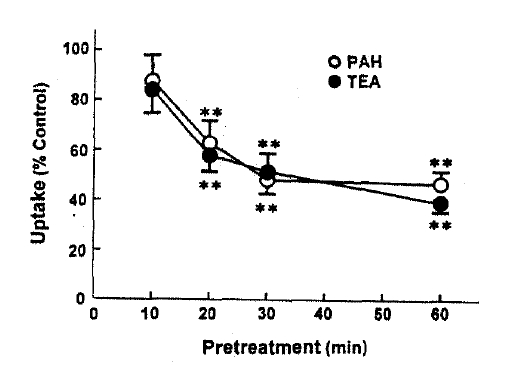
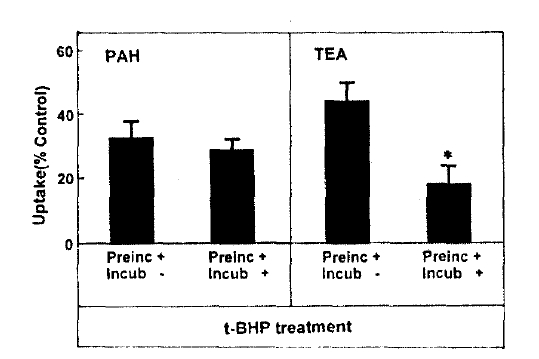
Fig. 2 depicts the effect of pretreatment time on t-BHP inhibition of organic ion uptake, slices were pretreated with 0.5mM t-BHP during 10ŌĆō60min and then organic ion uptake was measured for 60min. The inhibitory effect of t-BHP on PAH and TEA uptake increased with increasing pretreatment time by 30min. When the pretreatment time was extended to 60min, PAH uptake was not different from that obtained anfter 30min of pretreatment. Thus, subsequent experiments were perflormed after pretreating with t-BHP for 30min. In order to determune whether the effect of t-BHP was irreversible, slices were pretreated with 0.5mM t-BHP for 30min and then a 60-min organic ion uptake was measuredin the incubation medium with or without 0.5mM t-BHP. The results were summarized in Fig. 3. PAH uptake was not diferent between slices incubated with and without t-BHP, indication irreversible inhibition. On the other hand, TEA uptake was more depressed in both preincubation and incubation media with t-BHP, compared to that in the incubation medium without t-BHP.
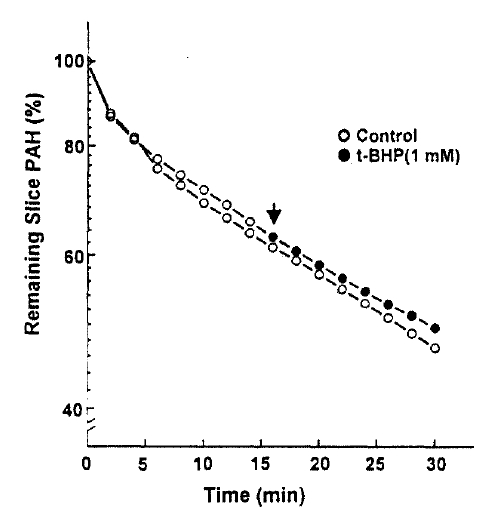
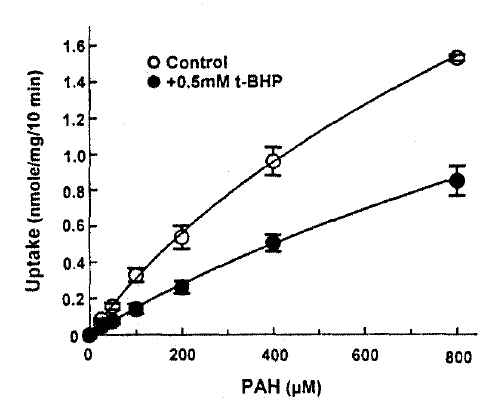
The efflux of PAH was measured in the presence and absence of 1mM t-BHP in the medium. The efflux of PAH was not altered by 1mM t-BHP(Fig. 4), and the efflux rate constant was 0.0176┬▒0.0043 and 0.0183┬▒0.0031 per min. in the presence and absence of t-BHP, respectively. This indicates that the inhibitory effect of t-BHP on the steady-state accumulation of PAH ins due primarily to the reduction in the influx of PAH across the basolateral membrane. In the next series of experiments, the kinetic analysis was performed to identify the nature of inhibition of t-BHP on PAH uptake. The rate of PAH uptake was determined during a 10-min incubation at barious PAH concentrations(25ŌĆō800╬╝M) in the slices pretreated with or without 1mM t-BHP for 30min. The results are depicted in Fig. 5. Analyses of teh data using a computerized model of Michaselis-Menten were performed to obtain the kinetic parameters. The results indicated the t-BHP resulted in a significant reduction in Vmax from 1.54┬▒0.74 to 0.72┬▒0.54╬╝mole/g/10min(p < 0.05), while it had no effect on Km for PAH(0.56┬▒0.24 vs. 0.69┬▒0.12mM, p < 0.1).
2. Effect of t-BHP Kon Oxygen Consumption
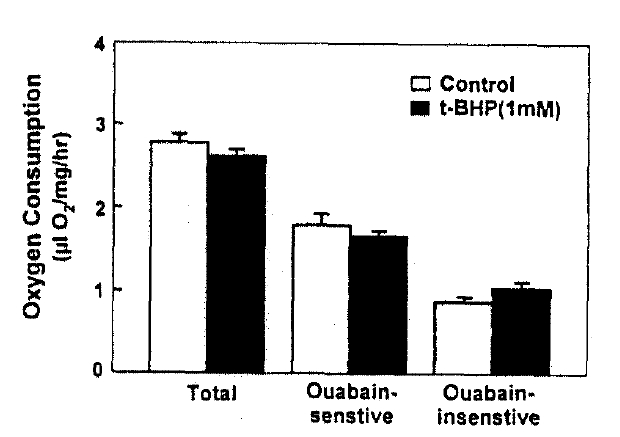
Since it has been reported that mitochondria are an important target of toxicity by t-BHP and other oxidant chemicals in the hepatocytes (Masaki et al.18), 1989; Nieminen et al.19), 1990; Redelged et al.20), 1990), oxygen comsumption was measured in the presence of 1mM t-BHP with or without 1mM ouabain; a specific inhibitor of Na+-K+-ATPase activity. The results are depicted in Fig. 6. Ouabain-senstive and -insenstive oxygen consumption were not changed by the treatment of t-BHP. These suggest that t-BHP did not impair the mitochondrial fuctkon or Na+-pump activity.
3. Effect of t-BHP on LDH Relesase
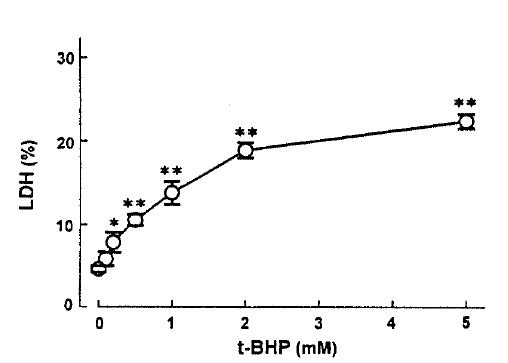
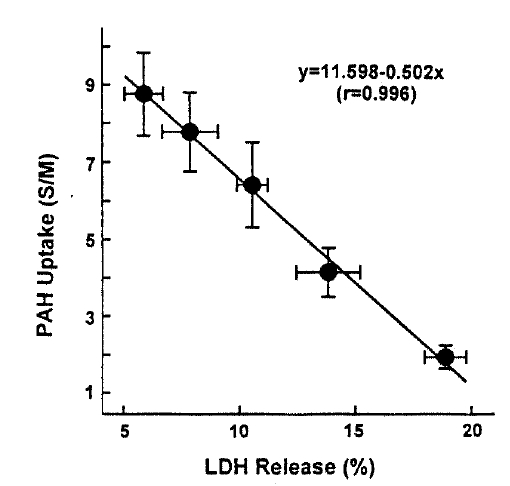
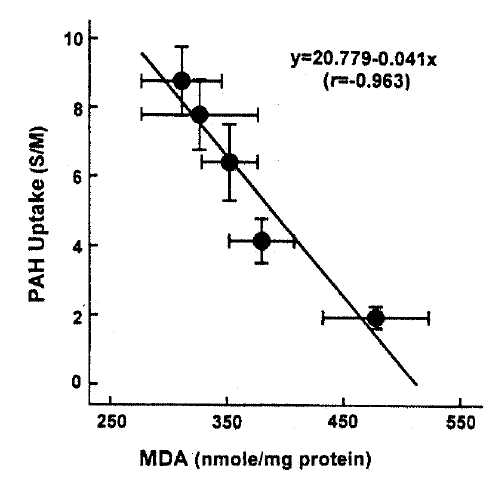
In an attempt to evaluate the effect of t-BHP on cell injury, the release ofLDHfrom slices into the medium was measured. When slices were incubated with t-BHP for 60min, t-BHP increased LDH release in a dose-dependent fashion over concentrations of 0.25ŌĆō2.0m(Fig. 7). There was no difference in LDH release between the 2.0 and 5.0mM t-BHP groups. The accumulation of PAH by renal cortical slices has been proposed as a sensitive indicator in the assessment of nephrotoxicity(Hirsch21), 1976). In the kidney, LDH release also has been used as an indicator for renal tubular cell damage(Bonventre et al.2), 1988). Therefore, in order to determine whether there is a correlation between the inhibition of PAH uptake by slices and the increase of LDH release, the uptake of PAH in the presence of t-BHP was plotted as fuction of the change in the release of LDH release was highly correlated with the reduction in PAH uptake.
DISCUSSION
Even though oxygen is absolutely required for aerobic life, it can also participate in potentially toxic reactions involving oxygen free radicals and transition metals such as Fe that damage membranes, proteins, and nucleic acids(Floys22), 1990). Under physiological conditions, oxygen free radicals are generated by autooxidation of a wide variety of small molecules or memlbrance-bound cytochromes and by numerous enzymes, localized in the cytosol, mitochondria, peroxisomes and plasma membranes(Freeman and Carpo23), 1982). In order to protect itself against exposure to oxygen free radicals, the eukaryotic cell has evolved oxygen free radical scavenging wystems which limit oxidative damage. However, when an excessive amount of oxygen free radicals are produced or antioxidative mechanisma are impaired, oxidative damage may occur and this appears to be important in contributing to several pathological conditions including ischemia-reperfusion injury(Bonventre et al.2), 1988). Since membranes possess polyunsaturated fatty acids most susceptible to oxidation(Polyer and McCay24), 1971), oxygen free radicals can induce lipid peroxidation which will cause changes in the structure of the membrane and thereby will result in changes in the activity of essential membrane proteins such as Na+-K+-ATPase (Floyd22), 1990). The results of the present study demonstrate that t-BHP, an exogenous oxidant, impaired accumulation of PAH and TEA in rabbit renal cortical slices. If t-BNHP effect on PAH efflux by the drug could be predicted. However, PAH efflux was not altered by 1mM t-BHP(Fig. 4), the concentration which reduced significantly PAH uptake(Fig. 1). This indicates that the inhibitory effect of t-BHP on the steady-state accumulation of PAH in the slices is not due to the increased efflux but due to the reduced influx of PAH from medium into the cell across the basolateral membrane. Interestingly, alterations in PAH uptake by t-BHP were highlyu correlated with the changes in LDH release in renal cortical slices(Fig. 7). LDH release has been widely used as an indicator for cell viability in various tissues including kidney tubules(Bonventre et al.2), 1988). Thus, these results suggest that the extent of cell injury could be assessed by measuring PAH uptake in cortical slices, as proposed by Hirsh21) (1976). In this study, the effect of t-BHP on PAH uptake was completely irreversible, but that on TEA uptake was partly reversible. This suggests that the action site of t-BHP on transport system for organic anion may be idfferent from that on organic cation transport system. Although it is highly speculative, it is possible that the location of PAH carrier in the membrance may be different from that of TEA carrier, resulting in variable dependence of their activity by t-BHP owing to a different lipid microenvironment.
The kinetic analysis showd that the Km for PAH influx was similar in the control and t-BHP-treated slices, indicating that substrate affinity of the carrier was not altered by t-BHP. However, the maximum rate of PAH influx(Vmax) appeared to be siginificantly reduced by t-BHP(Fig. 5). These results suggest that t-BHP causes a siginificant reduction in the number or the trurnover rate of active carriers.
Since it has been postulated that PAH transport across the basolateral membrane is coupled directly(Gerencser and Hong25), 1975) or indirectly (Pritchard & Miller26), 1991) to the Na+ gradient, t-BHP could reduce PAH uptake by inhibiting Na+-K+-ATPase activity. However, the functional Na+-pump activity(estimated as ouabain-sensitive oxygen consumption) was not altered by 1, M t-BHP, a concentration which inhibited PAH uptake by approximately 50%. It has been reported that mitochondria are an important target of toxicity by t-BHP and other oxidant chemicals in the hepatocytes(Masaki et al.18), 1989; Nieminen et al.19), 1990; Redelged et al.20), 1990). Both basal and ouabain-sensitive oxygen consumptions also decresase in renal proximal tubules treated with 1mM t-BHP for 30min(Borkan and Schwartz12), 1989; Schnellmann13), 1988). Whether t-BHP resulted in mitochondrial injury in this study is not clear. However, an eqaul rate of ouabain-sensitive oxygen consumption in the control and t-BHP-treated slices(Fig. 6) suggests that t-BHP did not impair the energy-producing catabolism linked to PAH influx.
This study demonstrates there is a correlation between PAH uptake and LDH release and lipid peroxidation in renal cortical slices subjected to various concentrations of t-BHP, suggesting that lipid peroxidation may play an improtnat role in reduction of PAH uptake by cortical slices is a senstive indicator in th assessment of nephrotoxicity, as indicated by Hirsch21) (1976). Lipid peroxidation by oxygen free radicals results in enhanced fragility of lipid-containing membranes (Thompson and Hess27), 1986). In this context, it is speculated that reduction in Vmax of PAH influx is attributed to lipid peroxidation in renal cortical slices. Peroxidative damage has been implicated in reducing D-gluocse transport primarily due to a decrease of Vmax while the affinity of substrate was not influenced in brush-border membranes isolated from kidneys(Molitoris and Kinne28), 1987) and intestine(Jourd'Heuil et al.29), 1993). However, the possibility is not excluded that t-BHP produces a direct injury in carrier protein for PAH transport. Im conclusion, t-BHP causes a significant inhibition of the uptake of PAH and TEA, which was accompanied by an increase in LDH release. Inhibition of PAH uptake was not due to altered cell metabolism. Exposure of slices to t-BHP, dose-dependent impairment in PAH uptake, was highly correlated with the increase in lipid peroxidation, suggesting that lipid peroxidation plays an important role in the inhibitory effect of t-BHP on PAH transport in rabbit proximal tubules.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





