


INTRODUCTION
The red blood cell (RBC) has several well characterized Na+ transport mechanisms including the Na+-K+ pump (ouabin-sensitive Na+ efflux), Na+-K+ cotransport (CoT: bumetanide-sensitive Na+ efflux) and passive Na+ permeabiluty (ouabain-and bumetanide-resistant Na+ influx). Among these Na+ transport mechanisms, the Na+-K+ pump is the main mechanism maintaining the Na+ and K+ diffusion.1)
Although a derangement of Na+ transport in chronic uremia has been reported, most studies have focused on the Na+-K+ pump identified by the membrane Na+-K+ ATPase enzyme system, with the general conclusion of suppressed enzyme activity in chronic uremia.2ŌĆō14) In recent studies, however, several reports observed a derangement of other Na+ transport pathways including Na+-K+ CoT, Na+-Li+ CTT and passive Na+ permeabilty in chronic uremia.15ŌĆō18) To carry out a further investigation of apparent change in the Na+ transport of RBC in patients with chronic uremia, the authors measured the Na+-K+ pump activity as well as Na+-K+ CoT, Na+-Li+ CTT, and Na+ passive permeability.
SUBJECTS AND METHODS
1. Subjects
Twelve male patients and eleven female patients with end stage renal disease (ESRD) receiving chronic maintenance dialysis were recruited for the study. Pertient clinical and pre-dialytic laboratory findings are depicted in Table 1. The age range of this study group was 27 to 66 (mean 49) years of age. The duration of maintenance hemodialysis was 7 to 120 (mean 35) months. The etiologies of ESRD and the number of patients, in parentheses, were as follows; chronic glomerulonephritis (sixteen), diabetic nephropathy (four), chronic pyelonephritis (one), polycystic kidney disease (one), and gouty nephropathy (one). Patients were dialyzed for 5 hours, 2 or 3 times weeklly using hollow fiber cuprophan disposable dialyzers. All patients were on acetate hemodialysis (dialysate composition in mEq/Ōäō 134, K+ 2.0, Ca++ 3.0, Mg++ 1.5, CIŌłÆ 105 CH3COOŌĆØ 35 and dextrose 2 g/l). The normal population studied consisted of 21 male and 26 female subjects with an age range of 24 to 37 years (mean: 30.2). All subjects were normotensive, and had no medical problems or family history of essential hypertension.
2. Methods
1) Preparation of RBC
Venuous blood (10ml) collected in heparinized tubes was centrifuged at 1750 g for 10 min, and the plasma and buffy coat were aspirated. RBC were then washed twice with isotonic MgCl2 (110 mM). All steps were carried out at 4┬░C.
2) Determination of Intracellular Na+ and K+
Cells were washed 3 times with 10 volumes of ice-cold isotonic MgCl2 solution. After mixing, duplicate hematocrit determinations were made of this suspension. Then 100 ╬╝Ōäō were diluted 1:100 in deionized water. The concentration of Na+ and K+ of the hemolysate was measured on a Perkin Elmer model 2380 atomic absorption spectrophotometer, and expressed as mmol/l of packed red cells by dividing the ionic concentration by the hematocrit and multiplying by the dilution factor.
3) Measurements of Ionic Fluxes
Simultaneous measurements of the Na+-K+ pump, Na+-K+ CoT, Na+-Li+ CTT and ouabain-and bumetanide-resistant Na+ and K+ fluxes (passive Na+ and K+ permeability) were made according to the modified method of Garay et al.19) Washed red cells were resuspended in cold Mg++ sucrose medium (mM); 75 MgCl2, 85 sucrose, 10 MOPS-Tris (pH 7.4 at 37┬░C) and 10 glucose, to a hematocrit of 20ŌĆō25%. A portion of the cell suspension was added (final hematocrit 4ŌĆō5%) to different cold solutions containing buffered Mg++ sucrose plus the following additions (mM): (1) 2 KCI, (2) 0.1 ouabain, (3) 0.1 ouabain plus 0.02 bumetanide, and (4) 10 LiCI. 0.02 bumetanide, and 0.1 ouabain. At time 0, the tubes were transferred to a 37┬░C bath for incubation. Sixty minutes after incubation, media 1, 2, 3 and 4 tubes were transferred in an ice bath for 1 min and then centriguged at 1,750g for 5 min at 4┬░C. The supernatant was carefully removed and the Na+ concentration was measured by atomic absorption spectrophotometry, using standards prepared in the appropriate Mg++ sucrose media.
Na+ and K+ effluxes were computed using the following equation:
D Cat ((╬╝mol/l supernatant) is the difference between the external cation concentration (Na+ or K+) after incubation at 37┬░C and that at 0 time. The Na+-K+ pump activity was calculated by subtracting the Na+ efflux in the presence (medium 2) from that in the absence (medium 1) of ouabain. The Na+-K+ CoT fluxes were obtained by subtracting the Na+ efflux in the presence of ouabain plus bumetanide (medium 3) from that in the presence of ouabain alone (medium 2), while for the calculation of Na+-Li+ CTT, Na+ and K+ effluxes in medium 3 was substracted from that in fluxes in medium 3 were taken as the ouabain-and bumetanide-resistant Na+ and K+ fluxes. The efflux rate constants for ouabainŌĆösensisitive, bumetanide sensitive, and passive Na+ and K+ effluxes, expressed in hŌłÆ1, were calculated as the ratio of the respective fluxes to the erythrocyte Na+ and K+ content.
RESULTS
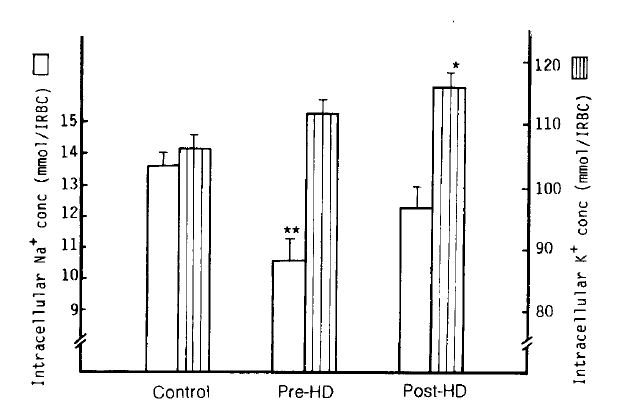
1. Intracellular Na+ and K+ Concentrations
The mean intracellular Na+ concentration [Na+]i value in the pre-dialytic group was significantly lower than that of the control group (10.6 ┬▒ 0.7 vs. 13.6 ┬▒ 0.4 mmol/l RBC; p<.001), but not statistically different from the post-dialytic group (12.3 ┬▒ 0.7 mmol/l RBC). The mean [K+]i value in the post-dialytic group was markedly increased over that of the control group (115.9 ┬▒ 2.1 vs. 105.8 ┬▒ 2.1 mmol/l RBC; p<.001), but there was no significant difference in the pre-dialytic group (11.5 ┬▒ 2.1 mmol/l RBC). The authors did not observe any significant correlation between the change of Na+-K+ pump (r = 0.06), the change of rate constant for ouabain-sensitive Na+ efflux (r = ŌĆō0.21) in the post-dialytic group and the decrease of body weight after hemodialysis (Table 2, Fig 1).
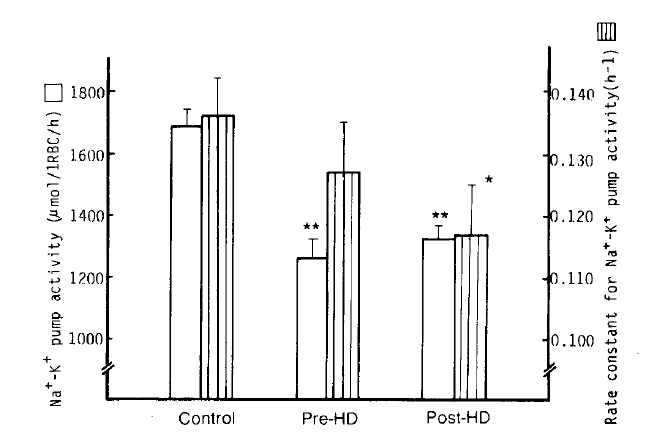
2. Na+-K+ pump (Ouabain-sensitive Na+ efflux)
It was found that the Na+-K+ pump activity of erythrocytes in the pre-and post-dialytic groups markedly decreased compared to that of the normal control group with statistical significance (1,263 ┬▒ 56 vs. 1,328 ┬▒ 42 vs. 1,688 ┬▒ 52 ╬╝mol/l RBC/h; p<.0001, respectively). The Na+-K+ pump activity in the post-dialytic group, however, tended to recover, with no significant difference. The rate constant for ouabain-sensitive Na+ efflux in the post-dialytic group was significantly lower than that of the normal control group (0.1167 ┬▒ 0.0075 vs. 0.1359┬▒0.0059 hŌłÆ1; p<.05), but not different from the pre-dialytic group (0.1265 ┬▒ 0.0077 hŌłÆ1) (Table 2, Fig 2).
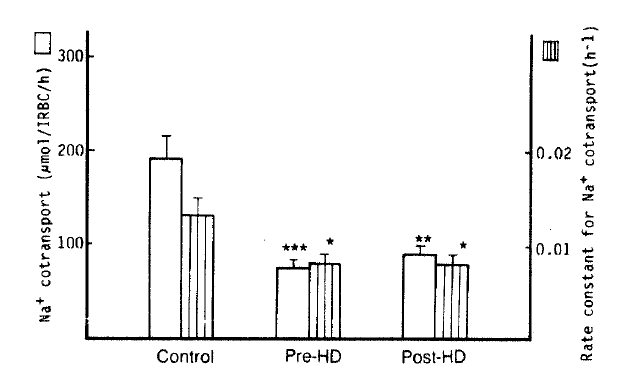
3. Na+-K+ Cotransport (Bumetanide-sensitive Na+ efflux)
The authors observed a significant decrease of Na+ CoT in the patients with pre- and post-dialytic uremia than that in normal subjects (75 ┬▒ 9 vs. 92 ┬▒ 9 vs. 192 ┬▒ 23 ╬╝mol/l RBC/h; p<.001, respectively). Also, the rate constants for Na+ CoT in the pre- and post-dialytic groups was markedly lower than that in the control group (0.0077 ┬▒ 0.0011 vs. 0.0079 ┬▒ 0.0009 vs. 0.0134 ┬▒ 0.0022 hŌłÆ1; p<.05 respectively). The K+ CoT in pre- and post-dialytic uremia was significantly lower than that in the control group (116 ┬▒ 15 vs. 102 ┬▒ 17 vs. 253 ┬▒ 28 ╬╝mol/l RBC/h; p<.01, 001, respectively) (Table 2, Fig. 3).
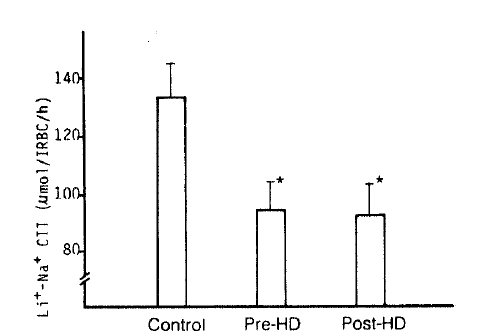
4. Na+-Li+ Countertransport (Li+-stimulated Na+ efflux)
The authors observed a marked decrease of the Na+-Li+ CTT value in the pre- and post-dialytic groups compared to that of the control subjects (94 ┬▒ 10 vs. 92 ┬▒ 11 vs. 133 ┬▒ 12 ╬╝mol/l RBC/h; p<.05, respectively). But, there was no significant difference between the pre-dialytic and post-dialytic groups (Table 2, Fig. 4).
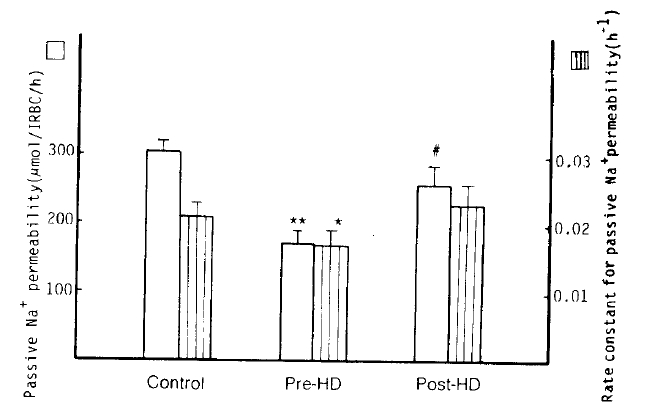
5. Passive Na+ Permeability (Ouabain and bumetanide-resistant Na+ influx)
Passive Na+ permeability in the patients with pre-dialytic uremia was markedly decreased as compared to the control group (173 ┬▒ 16 vs. 304 ┬▒ 16 ╬╝mol/l RBC/h; p<10ŌłÆ5); however in the post-dialytic group it tended to recover to the normal value, without a significant difference from the normal control group (259 ┬▒ 29 ╬╝mol/l RBC/h; NS). Also, the rate constant for passive Na+ permeability in the pre-dialytic uremia group was significantly lower than that in the control subjects (0.0171 ┬▒ 0.0020 vs. 0.0214 ┬▒ 0.0014 hŌłÆ1; p<.01). Passive K+ permeability in the pre-dialytic group was noticeably lower than in the normal group (1,558 ┬▒ 67 vs. 1,948 ┬▒ 68 ╬╝mol/l RBC/h; p<.001), but in the post-dialytic group it tended to recover to the normal value with significant difference from that of the pre-dialytic subjects (2,308 ┬▒ 208 ╬╝mol/l RBC/h; p<.01) (Table 2, Fig. 5).
DISCUSSION
Sodium movements across human RBC membranes are mediated by several transport systems. The ouabain-sensitive Na+-K+ pump is the main mechanism maintaining the Na+ and K+ electrochemical gradients across RBC membranes against the passive Na+ and K+ diffussion. At least three additional transport systems have recently been described in human RBC membranes: 1) Na+-K+ CoT; 2) Na+-Li+ CTT: 3) passive Na+ permeability1)
A derangement of ion transport in chronic uremia has been demonstrated in many tissues. An increase in [Na+]i was first observed in erythrocytes from patients with chronic renal failure over two decades ago2). Thereafter, many investigators concluded from their data that significantly high [Na+]i in erythrocytes came from uremic subjects4,5,6,8,12) Neverthless, in contrast to these results, others observed a decrease in [Na+]i of freshly drawn RBC from uremic subjects, although a significantly decreased Na+-K+ pump activity in uremic RBC was also observed by these individuals9,11,18) Corry et al., found that the [Na+]i in RBC from dialysis patients was similar to that of normal subjects17). In our study, the mean [Na+]i value in pre-dialytic subjects was significantly lower as compared with that in normal subjects, and in the post-dialytic group the [Na+]i tended to recover to the normal value. What is the possible mechanism for changes in the level of erythrocyte sodium from uremic subjects? Earlier efforts to explain the mechanism of the rise in [Na+]i were limited to measurements of pump-mediated Na+ efflux, the passive Na+ leak, and Na+-K+ ATPase activity2ŌĆō14.
Subsequently, several groups of investigators have provided evidence for a defect in the Na+ and K+ transport of erythrocytes2,4ŌĆō6,8ŌĆō14,20, leukocytes7), skeletal muscles21), intestine22) and brain23) in uremic subjects as well. However, despite the potential importance of a defect at the level of Na+-K+ ATPase in the genesis of uremia, a clear understanding of the nature of the abnormality and its correction by hemodialysis has not emerged. At least two reasons for this are apparent. First is the heterogeneity of the patient groups; patients with varying degrees of renal allure have been stuided, both before and after institution of dialysis. Second, and a more subtle problem, has been the heterogeneity of the methods used to study the Na+-K+ ATPase. This problem arises from the fact that there are several different ways in which the Na+-K+ ATPase can be probed, including measurement of (a) the specific binding of the inhibitory ligand [3H] ouabain, which can be used to determine the number of Na+-K+ pump units; (b) the activity of membrane Na+-K+ ATPase as a function of varying concentrations of substrates and ions, which measures the enzymatic function of the protein; and (c) the rates of specific Na+ and/or K+ across cell membranes, which assess the inhibit the Na+-K+ pump of erythrocytes.13,14,16,28) According to several researchers, OLF has been variably known as endigin, endoxin, endogenous digitalis, and putative natriuretic factor. Although OLF is not definitely clarified yet, it is described as having a low molecular weight of less than 500 daltons25), and by another author, as having a high molecular weight of 1,500ŌĆō10,000 daltons26). OLF, which is readily solule in an organic solvent and is heat-stable, is a peptide digested by prolidase, but not by trypsin and other enzymes. Its release from the hypothalamus can be detected by the increase in amount of extracellular fluid such as that which occurs in chronic uremics, water immersion, primary hyperalodosteronism and high salt loading. This OLF acts on the renal collecting ducts and tubules by inhibiting Na reabsorption, and its action causes a strongly enhanced natriuresis and diuresis24ŌĆō29).
However on the other hand, OLF inhibits the efflux of Na+ from the cell membrane by acting on the erythrocytes, leukocytes, platelets, nerve cells and smooth muscle cells of the vessel. In other words, OLF may partially inhibit the Na+-K+ pump in vascular smooth musclle, thereby causing an increase in norepinephrine secretion from nerve cells, and in intracellular Na+ ion concentration. Then, because the vascular smooth muscle cells contain a Na+-Ca++ exchange transport system in their plasma membrane, more free cytosolic calcium ions than normal are delivered to these cells. This causes an increased vascular tone and peripheral vascular resistance that elevated the blood pressure30). Welt (1969) and Yoon et al. (1986) had observed that erythrocyte Na+-K+ ATPase activity was markedly decreased when uremic plasma was incubated with erythrocyte membranes prepared from normal subjects4,14). Some investigators had observed the increase of Na+-K+ pump activity after dialysis, with a significant correlation to the amount of fluid removed. These observations are consistent with the hypothesis of the presence of OLF, i.e. a circulating Na+-K+ pump inhibitor in uremic plasma which is acutely influenced by volume change; predialytic hypervolemia leads to the release of this OLF, and its secretion is then inhibited by volume removal during hemodialysis13ŌĆō16) However, in our study, the Na+-K+ pump activity in the pre-dialytic group tended to recover after dialysis, but with no significant difference. Also, the authors did not find any significant correlation between the change of the Na+-K+ pump, the change of rate constant for ouabain-sensitive Na+ efflux in the post-dialytic group and the decrease of boody weight after hemodialysis. In the second place, other possible mechanisms of inhibition of the cell membrane Na+-K+ pump in chronic uremia can be considered to lead to uremic toxins (phenolic acid and methylguanidine), metabolic acidosis or vanadate within the body5,31).
The authors observed a significant inhibition of the bumetanide-sensitive Na+ efflux value (p<.00l, respectively) and rate constant for bumetanide-sensitive Na+ efflux (p<.05, respectively) in the patients with pre- and post-dialytic uremia than that of normal subjects, which reflected a new abnormality in the regulation of cellular Na+ homeostasis in uremia. The cause for reduction of bumetanide-sensitive Na+ efflux in chronic uremia is yet uncertain. Although there is a possibility of the presence of an endogeneous ŌĆśfurosemide-likeŌĆÖ factor proposed by Quarello et al.16), which is triggered by the extracellular volume expansion such as in pre-dialytic uremia, the authors have not seen a clear-cut correction of the Na+ CoT system after hemodialysis as shown in their report. The authorŌĆÖs finding is consistent with that of Corry et al.17), and Diez et al.18). The authors also observed a significant decrease of bumetanide-sensitive K+ efflux values in the patients with pre- (p<.01) and post-dialytic uremia (p<.001 versus those in normal subjects. It has been suggested by Duhm and Gobel32) that the bumetanide-sensitive K+ efflux system participates in plasma K+ homeostasis. They demonstrated an inverse correlations between the extracellular K+ concentration and the activity of the bumetanide-sensitive K+ efflux system in extruding K+ from RBC. As noted by Corry et al.17), since the pre-dialysis serum concentration of K+ in our patients was 5.5 ┬▒0.6 mEq/l, it is possible that the significant reduction of Na+-K+ CoT activity may be a direct result of this mild hyperkalemia. The pathophysiologic significance of this marked suppression in Na+-K+ CoT activity is also uncertain. It is interesting to note that the bumetanide-sensitive Na+-K+ CoT mechanism probably also operates in the diluting segment of the nephron where bumetanide inhibition of Na+ transport is known to cause potent natriuresis. Our study, therefore, raises the question whether a similar reduction in Na+ + K+ CoT activity also occurs in the diluting segment of uremic nephrons, and if so, whether such reduction could account for the natriuresis observed in chronic uremia17).
Red blood cells loaded with Li+ activate a one-to-one exchange of intracellular Li+ for extracellular Na+ through a Li+-stimulated Na+-Na+ CTT system. Woods et al. concluded that in both normal and uremic patients, Na+Li+ CTT of the RBC is dependent on a dialyzable plasma factor. In renal failure patients, the removal of this factor by dialysis or ultrafiltration led to depression of Na+-Li+ CTT33). The authors found that Na+-Li+ CTT in uremic patients was significantly decreased compared to that in normal subjects. The authors also found that the mean value from pre-dialytic subjects was not different from post-dialytic subjects. Thus, our study did not agree with the conclusion proposed by Woods et al. concerning the dependence of the Na+-Li+ CTT rate, on the presence of a dialyzable plasma factor.
In contrast to findings from Quarello et al.16), the authors have found that the passive Na+ permeability in the patients with pre-dialytic uremia was markedly decreased compared to that in normal subjects (p<10ŌłÆ5), but its value in the post-dialytic group tended to recover to a normal level, which was consistent with a previous report by Diez et al.18). Although the exact mechamism for inhibition of passive Na+ permeability in chronic uremia is as yet uncertain, changes in the lipid composition and distribution of the RBC membranes, as Bleiber et al. previously reported in uremic patients, might be involved in the observed alteration in passive Na+ fluxes34).
Our studies demonstrate that another possible mechanism of inhibition of the Na+-K+ pump in pre-dialytic uremia might be a secondary adaptive response of the cell of maintain normal intracellular ion concentration and transmembrane ion gradients in the face of reduced [Na+]i due to decreased passive Na+ permeability. Thus, the inhibition of Na+-K+ pump activity in pre-dialytic uremia might be regarded as an adaptive cellular response to decreased [Na+]i due to passive Na+ permeability, rather than the pure effect of OLF, uremic toxins, etc. This finding also raises the possibility that a decrease in passive Na+ permeability might be a proximal event due to changes in the lipid composition of the RBC membranes.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





