


INTRODUCTION
Lymphomatoid papulosis (LyP) is an uncommon disorder which is characterized by waxing and waning papulonodular cutaneous lesions that last from 4 to 5 weeks and often heals with hypopigmented or hyperpigmented scaring1). LyP was first described by Macauley in 19682) and it was defined as a clinically benign but histologically maiignant process. About 10% to 20% of patients will have associated with lymphomas, including mycosis fungoides, T-cell immunoblastic lymphoma or HodgkinŌĆÖs disease, which can precede, occur simultaneously with or follow the diagnosis of LyP1, 3). Anaplastic large cell lymphoma (ALCL) is histologically and phenotypically similar to LyP and also appears as part of this disease spectrum. Diagnosis of ALCL is occasionally difficult when only histologic examination is used, and it is aided by immunohistochemical and molecular biology techniques. The analysis of immunophenotype and T-cell receptor genes has been used extensively to determine the lineage and clonality of lymphoid cell population. It suggests that LyP and ALCL are derived from an identical T-cell clone4ŌĆō7). We report a case of lymphomatoid papulosis associated with relapsed Ki-1 positive anaplastic large cell lymphoma after complete remission in a 60-year-old Korean man.
CASE REPORT
A 60-year-old Korean man was admitted to Soonchunhyang University Hospital because of fever and multiple skin eruptions with crusts for 3 days, which were pruritic and erythematous. Papulonodular lesions were located on the anterior chest wall.
There was a history of pulmonary tuberculosis up to 10 years ago, and he had taken drugs for a year. A year prior to admission, he had had a history of enlargement of both cervical lymph nodes and postprandial epigastric pain, which was diagnosed to be early gastric cancer, Ki-1-positive anaplastic lymphoma, stage IIIB involving right submandibular lymph nodes, left cervical lymph nodes and hepatoduodenal lymph nodes (Fig. 1) without bone marrow involvement. At that time, he was treated with 6 cycle combination chemotherapy of ProMACE-CytaBOM regimen for Ki-1-positive anaplastic lymphoma during 6 months. Subtotal gastrectomy and gastroduodenostomy were performed for early gastric cancer after 2 cycles of chemotherapy. After 6 cycle combination chemotherapy, he was on complete remission of the disease.
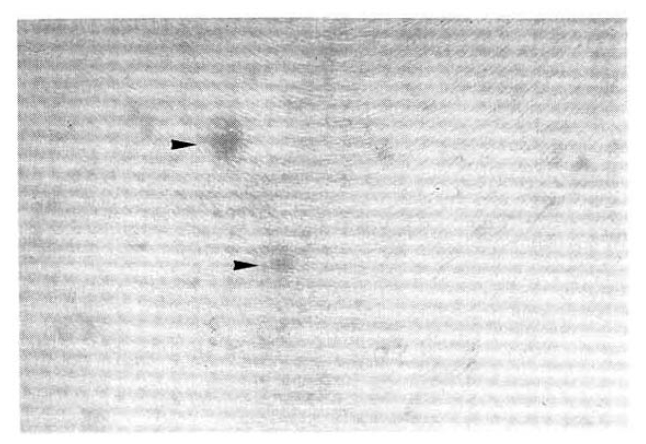
On admission, the temperature was 38.4┬░C, the pulse rate 85/min and the respiration rate 20/min. The blood pressure was 110/70mmHg. He had a chronic ill-looking appearance. The conjunctiva was pale and the sclera was not icteric. There were multiple small sized papulonodular eruptions on the anterior chest wall (Fig. 2). On examination of chest, crackles were heard on left lower lung field. Examinations of heart, abdomen and extremities were not remarkable. Enlargement of peripheral lymph nodes was not seen.
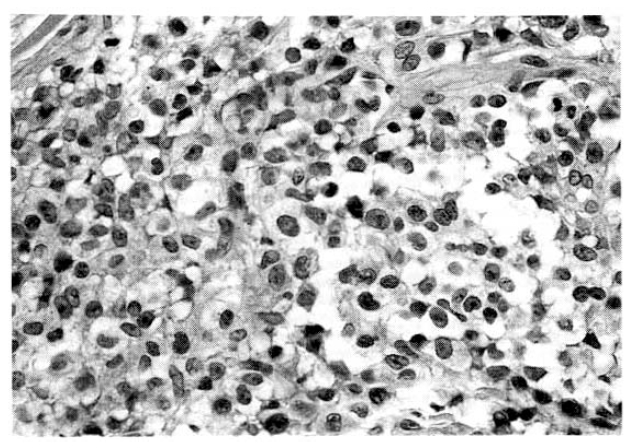
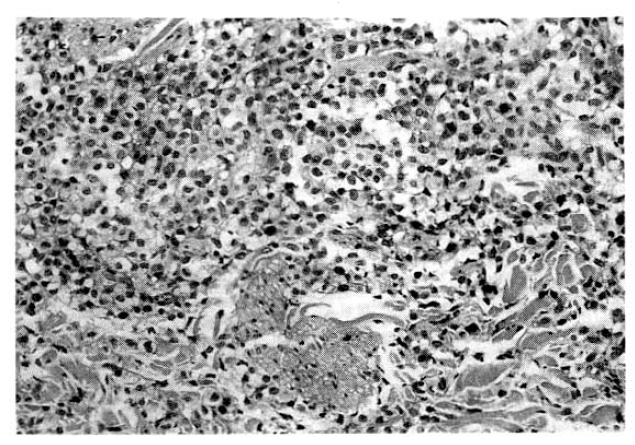
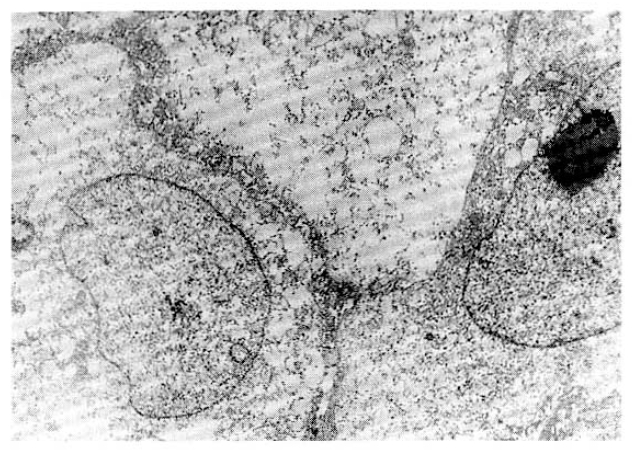
Laboratory evaluation revealed hemoglobin of 7.4g/dl, hematocrit of 22.4 percent, leukocyte of 14,600/mm3 with 66 percent neutrophils and 8 percent lymphocytes and platelet 300,000/mm3. The erythrocyte sedimentation rate was 71 mm/hour. Results of serum chemistry test were normal with exception of LDH 1003.3IU/I, BUN 29.9mg/dl, creatinine 1.9mg/dl. The electrocardiography was normal. Radiologic examination of chest revealed fibrostreaky densities in right upper lobe and CT scan of abdomen showed no intra-abdominal lymphadenopathy. Biopsy of cutaneous lesions on anterior chest wall and bone marrow aspiration and biopsy were performed. Light microscopic examination of skin specimen showed that the dermis was diffusely infiltrated with large pleomorphic tumor cells, which were strongly positive for Ki-1 immunohistochemical stain (Fig. 3, 4). Electromicroscopic examination revealed convoluted nuclei with prominent nuclei and sparse intracytoplasmic organelles (Fig. 5). Bone marrow aspiration and biopsy showed a focus of collection of atypical cells with complete necrosis, suspicious for involvement of malignant lymphoma. On diagnosis of relapsed Ki-1-positive anaplastic lymphoma stage IV, we performed salvage chemotherapy with cisplatin, dexamethasone, mitoxantrone and etoposide but, unfortunately, he died of persistent pancytopenia, followed by sepsis.
DISCUSSION
LyP is an uncommon cutaneous disorder composed of infiltrating clonal T-cells presenting as recurrent papulonodular eruptions7). The possible lymphoid origin of the large atypical cells (LAC) found in the dermis was elucidated by Stein et al.8) in 1985, using monoclonal antibody directed against the Ki-1 (CD30) antigen present on these cells. Recent reports, analyzing immunophenotype and T-cell receptor gene rearrangements in patients with both LyP and lymphoma, suggest that they are derived from an identical T-cell clone4ŌĆō7) and have a close relationship with the neoplastic process to malignant lymphoma. The association between lymphomatoid papulosis and malignant lymphoma is well known but still raises the problem of nosology between these two pathologies. Anaplastic large cell lymphoma (ALCL) is histologically and phenotypically similar to LyP and also appears as, part of this disease spectrum. The typical presentation of LyP is that of numerous small (smaller than 1cm) papules or nodules that may become hemorrhagic or centrally necrotic1, 9). These lesions resolve with hyperpigmented or hypopigmented scarring in 4ŌĆō6 weeks, and are rarely pruritic and typically involve the trunk or extremities, sparing the mucosa, face, palms and soles1, 9). In our case, the lesions are multiple pin head sized erythematous papules with crust formation in anterior chest wall.
Male: female ratios vary from 2:1 to 3:2 in the reported series10, 11). With respect to the epidemiology of LyP, patients have a significantly increased frequency of prior or coincident lymphoproliferative disorders and an increased frequency of nonlymphoid malignant lesions and prior radiation exposure. The significance of the last two associations is unclear, whereas the association with other lymphoproliferative disorders suggests a common unknown inciting event or immune defect12).
The proliferation is histologically malignant and may possess any or all of the features of T-cell malignancy, including aberrant T-cell antigen expression and clonal rearrangement of T-cell receptor genes. The component cells are often highly anaplastic and express the CD30 antigen. In such cases, histopathologic features considerably overlap those of ALCL. In other instances, the histologic features more closely resemble those of mycosis fungoides. The histomorphologic examination of LyP is characterized by the following features: wedge shape or perivascular dermal infiltrated, small lymphocytes surrounding peripheral blood vessels.
Two distinct histologic subtypes of LyP have been described, as initially reported by Willemze et al.13) in 1982. The LyP type A lesion is characterized by a predominance of CD30-positive large anaplastic cells with abundant cytoplasm, pale vesicular nuclei and prominent nucleoli with occasional multinucleated cells resembling the Reed-Stemberg cell in HodgkinŌĆÖs disease, and cannot be distinguished morphologically from ALCL. The LyP type B lesion is characterized by a population of CD30 negative mononuclear cells with hyperchromatic cerebriform nuclei similar to the Sezary cell of mycosis fungoides. This histologic divergence has been confirmed by T-cell receptor and immunoglobulin gene analysis, with Type A histology having an increased risk of developing into malignant lymphoma14, 15). Lesions of our case were thought to be LyP type A for histomorphology and immunohistochemical stain.
Beljaards and Willemze have reported the prognosis of patients with malignant lymphoma associated with LyP15). They reviewed 50 cases of LyP associated lymphoma (38% MF, 24% HodgkinŌĆÖs disease and 32% ALCL) and found benign clinical courses in patients with mycosis fungoides, HodgkinŌĆÖs disease and cutaneous ALCL. In contrast, patients with ALCL and extracutaneous disease did poorly, with six of nine dying at a median follow-up period of 3 months after diagnosis. In our case, he who had diagnoses of extracutaneous diseases at presentation showed poor response to chemotherapy. Fernando and James calculated the cumulative risk in their series and it was clear that the risk for transformation begins 5 years after the first sign of LyP and continues to increase with time16).
Treatment of LyP is supportive care with observation. Various modality, including low-dose methotrexate, tetracycline, oral psoralen with PUVA, acyclovir, nitrogen mustard, steroids, and radiation therapy were reported to be efficacious1, 16, 17ŌĆō19). Intensive combination chemotherapy appears to have only a temporary effect on the disease18).
Our patientŌĆÖs clinical courses serve to illustrate the similarities and differences found between the entities of LyP and ALCL. Because entities of these diseases are not clear cut, further research to identify nature, prognosis and treatment of LyP should be given high priority.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





