


INTRODUCTION
In 1947, Owren1) described a female patient with a hemorrhagic disorder due to previously unrecognized coagulation factor, and coined the term parahemophilia. Owren called this new coagulation factor discovery, ŌĆ£FactorŌĆØ V. The factor V deficiency shows usually autosomal recessive trait inheritance.
The hereditary factor V deficiency is very rare, and probably fewer than 500 patientŌĆÖs affected worldwide have been reported since its discovery in 19432). The incidence has been estimated to be less than 1 in 1 million. Heterozygotes are usually asymptomatic and intermediate or normal plasma factor V levels. Only homozygotes patients have hemophilia-like hemorrhages and markedly decreased plasma factor V levels2).
Hemorrhagic manifestations of the factor V deficiency were ecchymosis, epistaxis and bleeding of oral cavity. Spontaneous intracranial bleeding was uncommon and occurred in approximately 6 to 10 percent of factor V deficiency of patients3).
Most bleeding episodes in patients with factor V deficiency could be treated with sufficient fresh frozen plasma to raise the plasma factor V level to 30%2).
CASE REPORT
A patient, 53-year-old male, visited the emergency room of our university hospital on July 22 1995 for sudden onset of drowsy mental status. On admission, the blood pressure was 140/90mmHg, the pulse rate was 72/min, body temperature was 36.8┬░C and respiration rate was 20/min. He had had no diabetes mellitus, no hypertension, no hemorrhagic diathesis in past histories. He had no history of acute trauma.
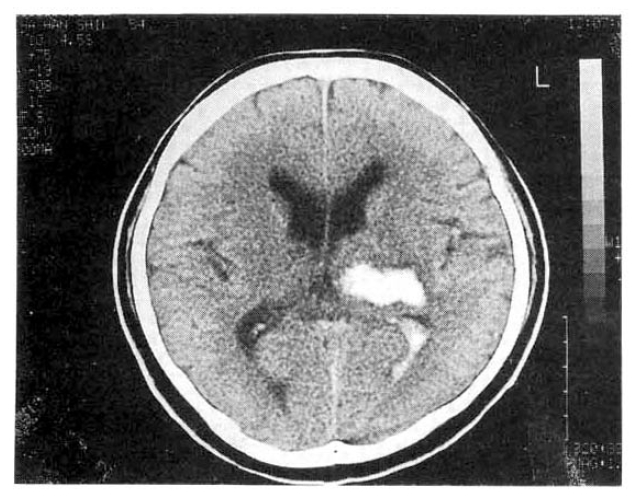
In neurologic examinations, he had a deep drowsy mental status, disconnected orientation, sensory dysphasia but intact cranial nerve system, motor and sensory systems. Reflexes were normal. Brain CT showed intracranial hemorrhage in left thalamus and intraventricular hemorrhage in occipital horn of Left lateral ventricle(Fig. 1).
Laboratory results as follows: CBC and ESR were normal. Bleeding time was normal (3 min). Both prothrombin time (PT 18.0 sec, INR 2.2, normal range < 1.14 INR) and activated partial thromboplastin time (APTT 49.0 sec, normal range < 35 sec), were markedly prolonged. Thrombin time was normal (5.4 sec; normal range 4┬▒2 sec). The levels of the coagulation studies on the patient revealed significant decrease of factor V (below 1%; normal range 60ŌĆō140%). Other coagulation factors were normal. After fresh frozen plasma was replaced, factor V recovered to 15%, prothrombin time recovered to 15.0 sec (INR 1.5) and activated partial thromboplastin time recoverd to 40.0 sec.
His parents had expired when he was young. He had five brothers and one sister. The were alive and had had no bleeding tendency. He had one son and three daughters. We examined the coagulation factors and PT, APTT of his offsprings. All their laboratory data were normal except for mild decrease of the concentration of factor V of his son (45%; normal range 60ŌĆō140%). His son had had no bleeding tendency.
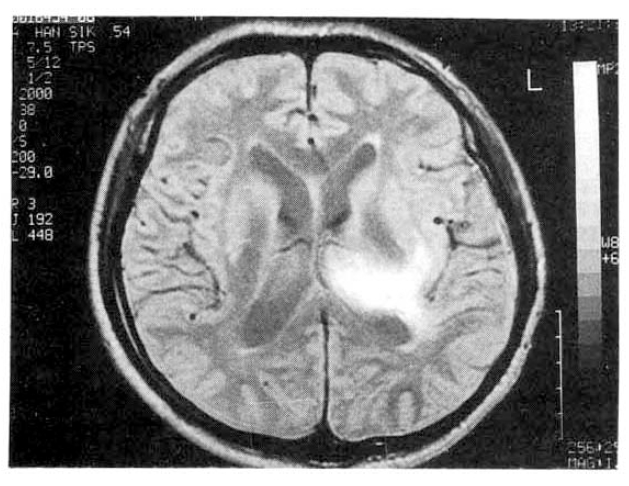
He recovered with fresh frozen plasma and supportive care. Two weeks later, follow-up brain MRI showed decreasing size of intracranial hemorrhage and stablized subacute hematoma (Fig. 2). He completely recovered after three weeks from admission day and has been followed at the out-patient clinic.
DISCUSSION
In 1944, Owren described a patient who had a bleeding diathesis attributable to the lack of single blood clotting factor, namely factor V or labile factor1). Owren designated the disease ŌĆ£parahemophilia,ŌĆØ to emphasize the clinical similarity to hemophilia. At that time, only four coagulation factors were considered: fibrinogen, calcium, prothrombin, thrombokinase. After owren reported parahemophilia, it was soon found that labile factor and factor V were one and same factor3).
At present, characteristic features of coagulation factor V established molecular weight: < 700kd4), plasma concentration: 7╬╝g/mL5), biologic half time: 12ŌĆō15 hour3). Factor V synthesis has been demonstrated in the hepatoma cell line HepG2 and human megakaryocyte and bovine aortic endothelial cells6, 7). Production of plasma factor V appears to occur primarily in the liver. The liver, as the source of plasma factor V, consisted of whole organ perfusion experiments showing factor V production by the liver. Biosynthesis of factor V that are structurally, functionally and immunologically indistinguishable from plasma factor V has been demonstrated in a human hepatocellular carcinoma cell line, HepG2, suggesting that the hepatocyte is the site of synthesis within the liver6). Another major site of factor V production was the megakaryocyte. In 1982 Tracy PB et al. found that platelets synthesized factor V. They reported that normal range of factor V concentration was from 4 to 14╬╝g/mL of plasma, while washed platelets indicated that 0.63 to 1.93╬╝g/mL of factor V was present per 2.5├Ś108 platelets. When corrected to individual hematocrits and platelets count, the data indicated that platelets contributed approximately 18% to 25% factor V of whole blood7). Factor V is stored in the platelet ╬▒ granules and is released during activation8). Bleeding diathesis and the concentration of plasma factor V in patients with factor V deficiency was not as well related as plasma factor V level3). Miletich et al. have proposed that platelet factor V level as much better correlated with bleeding symptoms9).
In contrast, the other coagulation factors that function as enzymatically active serine protease activators, factor VIII and factor V, are cofactors in the coagulation cascade. Activated factor V is a cofactor in the conversion of prothrombin to thrombin by factor Xa in an action that are phospholipid and calcium dependent. The activator complex formed by factor Xa, activated factor V, phospholipid and calcium was called the prothrombinase complex10, 11). Factor V was converted to factor Va by thrombin generating a heavy chain and light chain, and these two chains were held together by calcium ions. A connection, originally located between the heavy and light chains, is liberated during the active reaction4). Activation of factor V by thrombin is achieved through a series of proteolytic cleavages. As a result of these cleavages, single chain precursor factor V consisted of a 115-kd heavy chain and 73-kd light chain4, 12). Activated factor V was inactivated by activated protein C in a surface dependent reaction that requires calcium ions and nonenzymatic cofactor and protein S13).
Factor V deficiency was diagnosed by specific factor V assay. Since factor V acts as a cofactor in both intrinsic and extrinsic coagulation pathways, screening tests typically revealed prolongation of prothrombin time and partial thromboplastin time and normal thrombin time3). Factor V deficiency was usually considered to be inherited in autosomal recessive manner, although some authors considered it to be partially dominant14).
Only homozygous patients had hemophilia-like hemorrhage or thrombotic manifestations, while heterozygotes were usually asymptomatic and intermediate or normal plasma factor V levels15). Because his parents had died early, our patient had not typical autosomal recessive inheritance trait. The factor V of his son was mild decreased; it was probably subclinical deficiency. It suggested that he might be homozygous and his son heterozygous.
The predominant symptoms of factor V deficiency included ecchymosis, epistaxis and particular menorrhagia. Deep intramuscular hematoma and hemarthrosis were infrequent3). Epistaxis, an almost uniform problem, could be easily controlled with nasal packing and transfusion of fresh frozen plasma3). In the female, menorrhagia, which was a serious cause of death with the first menstrual period, had been reported in factor V deficiency16). Excessive bleeding occurred following dental extractions, trauma and surgery, but it is milder and much better controlled than the bleeding associated with hemophilia A3).
Hematuria, gastrointestinal bleeding or hemarthrosis occurred in approximately 15 to 20 percent3). Our patient had no ecchymosis, epistaxis, oral bleeding and other bleeding manifestations. He developed only spontaneous intracranial hemorrhage. Spontaneous intracranial hemorrhage occurred in approximately 6 to 10 percent of factor V deficiency patients3). A patient with factor V deficiency with intracranial hemorrhage was first reported in 1947 and manifestated as blindness1). Usually, factor V deficiency with intracranial hemorrhage developed during prenatal and childhood period17, 18). Adult onset of spontaneous intracranial hemorrhage was very rare.
Although factor V deficiency is a milder disorder than hemophilia A, patients with factor V deficiency may sometimes require replacement therapy. Treatment is usually with fresh frozen plasma, since no commercial concentrates of factor V are available. Platelet concentrates may also provide hemostasis in patients with factor V deficiency. The level of factor V activity in the patientsŌĆÖ plasma is usually < 5% of that in normal plasma19). Most bleeding episodes in patients with factor V deficiency can be treated with sufficient fresh frozen plasma to raise the plasma factor V level to 30%2).
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





