


INTRODUCTION
Anti-platelet therapy is fundamental for primary and secondary prevention of cardiovascular diseases [1,2]. Aspirin and clopidogrel are the most widely used anti-platelet agents in cardiovascular diseases. Current guidelines recommend aspirin and P2Y12 inhibitors for patients with coronary stent implantation for designated periods and permanent aspirin thereafter [3]. However, many patients still experience adverse ischemic events even during dual anti-platelet therapy, suggesting unmet needs of novel anti-platelet effects without increasing bleeding risk. About 5% to 45% of patients with aspirin and 5% to 30% of patients with clopidogrel showed significant residual platelet aggregation activities [4-6], and those who show resistance to the current standard anti-platelet agents revealed poor clinical outcome [7]. Moreover, recent development of new potent anti-platelet agents such as prasugrel and ticagrelor elicited the concern for hemorrhagic side-effects. Therefore, development of new anti-platelet agents with stable anti-platelet effect and minimal bleeding risk would be of great value in patients with cardiovascular diseases [8].
Epigallocatechin-3-gallate (EGCG), which is extracted from green tea, has been recently demonstrated to have anti-platelet effect in addition to anti-oxidative, anti-angiogenic, and anti-cancer effects [9-12]. EGCG also improved platelet concentration preservation through the inhibition of platelet activation and apoptosis [13]. However, there were limited reports of EGCG on detailed platelet functions especially in participants with anti-platelet agents. In this study, we explored in vitro anti- platelet effects of EGCG on human platelets obtained from the participants under the current anti-platelet therapy.
METHODS
Study design
From November 2014 to March 2015, we recruited the participants from the outpatient clinic of Cardiovascular Center in Korea University Anam Hospital. After the clinical evaluations, the participants with prior history of myocardial infarction, stroke, percutaneous coronary intervention and coronary artery bypass graft, clinical evidence of myocardial ischemia documented by stress tests, presence of angiographically significant (Ōēź 50% of diameter stenosis) coronary artery stenosis, and taking other anti-platelet agents were excluded. Finally, 40 participants were randomized into four groups: (1) control group (no anti-platelet agent), (2) aspirin group (ASA; aspirin 100 mg once daily), (3) clopidogrel group (CPD; clopidogrel 75 mg once daily), and (4) ticagrelor group (TCG; ticagrelor 90 mg twice daily). Participants of each group except the control group were administered with the randomized anti-platelet agent for 7 days. The study was approved by the Institutional Review Board of Korea University Anam Hospital (IRB No. 14130) and performed in accordance with the principles of the Declaration of Helsinki. Written informed consents were obtained.
Preparation of EGCG
EGCG was obtained from Sigma-Aldrich (St. Louis, MO, USA; Cat. No. E4268), and stock solution was prepared in dimethyl sulfoxide as 10 mM concentration.
Blood preparation
All blood samples were obtained in the morning after at least 8 hours of overnight fasting and drawn in tubes containing 3.2% sodium citrate. Anti-coagulated whole blood was used in this study. The isolated whole blood was incubated with either EGCG or control for 30 minutes at 37┬░C.
Multiple electrode platelet aggregometric assay
The extent of platelet aggregation was assessed using Multiplate Analyzer (Roche Diagnostics, Mannheim, Germany) as previously described [14]. Briefly, 300 ╬╝L whole blood was mixed with 300 ╬╝L 0.9% normal saline and stirred for 3 minutes at 37┬░C. Various concentrations of EGCG were preincubated with whole blood for 30 minutes at 37┬░C, and then the mixed blood was tested with Multiplate Analyzer. Adenosine diphosphate (ADP, 6.5 ╬╝M), arachidonic acid (AA, 0.5 mM), collagen (COL type, 3.2 ╬╝g/mL), or thrombin receptor activating peptide (TRAP, 32 ╬╝M) was added, and platelet aggregation was continuously recorded for 6 minutes. The adhesion of aggregated platelets to the electrodes led to an increase of impedance, which was detected for each sensor unit separately and transformed to aggregation units (AU) against time.
Shear stress-induced platelet adhesion assay
Shear stress-inducing platelet adhesion was analyzed by the Impact-R system (Matis Medical Co., Beersel, Belgium) as previously described [15]. Briefly, based on the cone and platelet analyzer technology, 130 ╬╝L whole blood was placed in a polystyrene well and set to flow (1800 S-1) using a rotating cone. After 2 minutes, the well was gently washed with PBS and stained with the May-Gruenwald stain. Platelet deposition was measured as the surface coverage (SC) and average size (AS) of the adherent platelet particles by the Impact-R image analyzer system connected to a microscope.
Flow cytometric analysis
The expressions of P-selectin and PAC-1 on the surface of platelets were measured by flow cytometry (FACS Calibur, Beckton Dickinson, San Jose, CA, USA) calibrated daily with Spherotech RCP-30-5 calibration beads (Sphereotech, Lake Forest, IL, USA) to assure consistent fluorescence measures [16]. Commercially available monoclonal antibodies such as fluorescein isothiocyanate (FITC)-conjugated PAC-1 (for the detection of activated GPIIb-IIIa; Becton Dickinson, San Jose, CA, USA), phycoerythrin (PE)-conjugated anti-P-selectin (Beckton Dickinson), and CD41a-Percp-Cy5.5 (for the identification of platelets; Becton Dickinson) were used in the analysis. In brief, 15 ╬╝L whole blood was incubated with these antibody mixture for 15 minutes at room temperature. Platelet P-selectin and PAC-1 were expressed as mean fluorescent intensity (MFI). A total 10,000 events were acquired and analyzed for the data analysis.
Statistical analysis
Categorical variables were reported as count and continuous variables as the mean ┬▒ standard error (SE). Independence of categorical variables was analyzed by the chi-square test. Multiple comparison was performed by using Tukey methods. Continuous variables were analyzed by the 1-way ANOVA. Only p values less than 0.05 were considered statistically significant. All statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).
RESULTS
Baseline demographic characteristics are shown in Table 1. There was no significant difference among four groups except that the mean age was higher in the CPD group than the other groups (Control, 52.0 ┬▒ 15.2 years; ASA, 53.9 ┬▒ 12.0 years; CPD, 66.5 ┬▒ 7.3 years; TCG, 58.3 ┬▒ 8.5 years; p = 0.03).
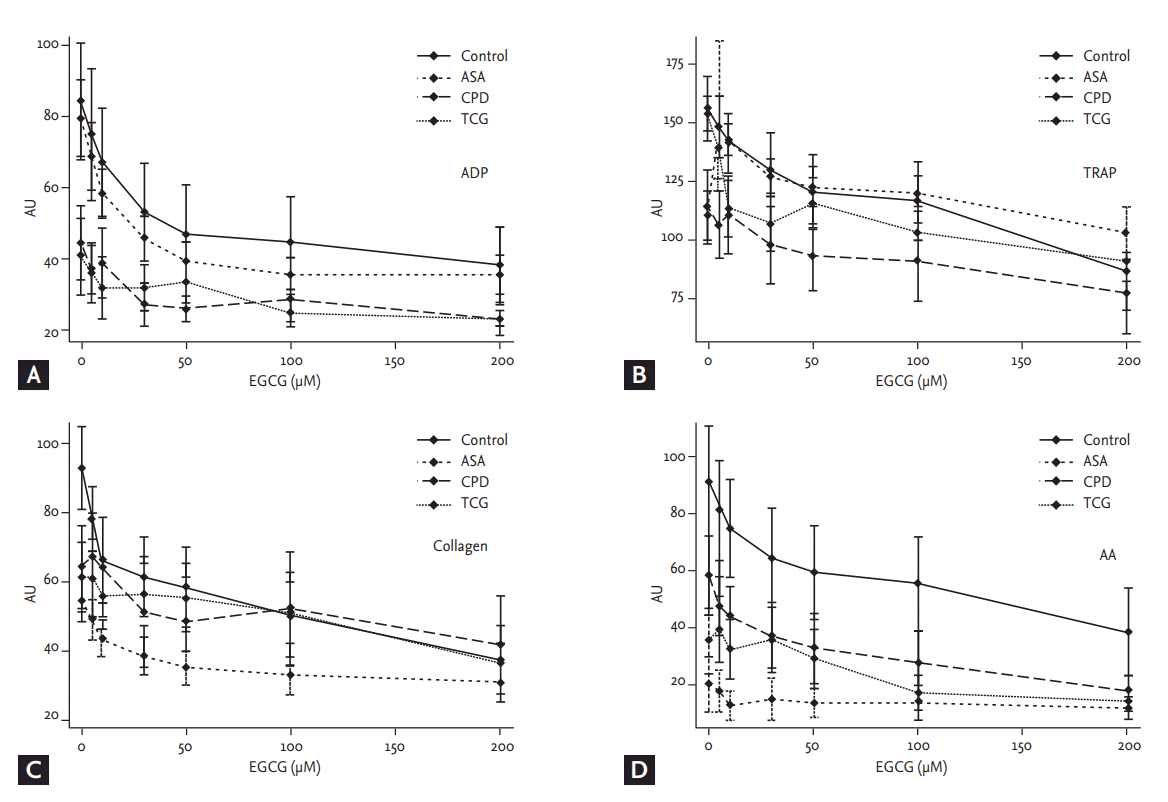
Agonist-induced platelet aggregation
Basal ADP- and TRAP-induced platelet aggregations were lower in the CPD and the TCG groups (ADP: Control 41.1 ┬▒ 27.5, ASA 38.7 ┬▒ 17.9, CPD 21.8 ┬▒ 16.1, TCG 18.1 ┬▒ 17.3, p < 0.01; TRAP: Control 98.5 ┬▒ 27.7, ASA 94.0 ┬▒ 16.6, CPD 73.5 ┬▒ 30.6, TCG 70.4 ┬▒ 21.4, p = 0.03) (Fig. 1). AA-induced platelet aggregation was lower in the ASA group (Control 50.9 ┬▒ 38.34, ASA 9.3 ┬▒ 17.1, CPD 30.1 ┬▒ 24.1, TCG 18.1 ┬▒ 21.9, p < 0.01), whereas COL-induced platelet aggregations were comparable among four groups.
EGCG significantly reduced ADP- and COL-induced platelet aggregation in a dose-dependent manner in the Control group (ADP, p = 0.04; COL, p < 0.01). However, EGCG showed only marginal inhibitory effects on TRAP- and AA-induced platelet aggregation in the Control group (TRAP, p = 0.10; AA, p = 0.10) (Fig. 1).
In the ASA group, EGCG significantly reduced ADP-, TRAP-, and COL-induced platelet aggregation in a dose-dependent manner (ADP, p < 0.01; TRAP, p < 0.01; COL, p = 0.04). However, EGCG did not additionally reduce AA-induced platelet aggregation in the ASA group (p = 0.94) (Fig. 1). In the CPD group, EGCG did not significantly reduce ADP-, TRAP-, and COL-induced platelet aggregation in a dose-dependent manner (ADP, p = 0.28; TRAP, p = 0.31; COL, p = 0.12). EGCG marginally reduced AA-induced platelet aggregation in the CPD group (p = 0.05) (Fig. 1). In the TCG group, EGCG did not significantly reduce ADP-, TRAP-, COL-, and AA-induced platelet aggregation in a dose-dependent manner (ADP, p = 0.82; TRAP, p = 0.14; COL, p = 0.58; AA, p = 0.37). There was no significant difference in ADP-, TRAP-, AA-, and COL-induced platelet aggregation among four groups under the maximal EGCG treatment (200 ╬╝mol/L) (Fig. 1).
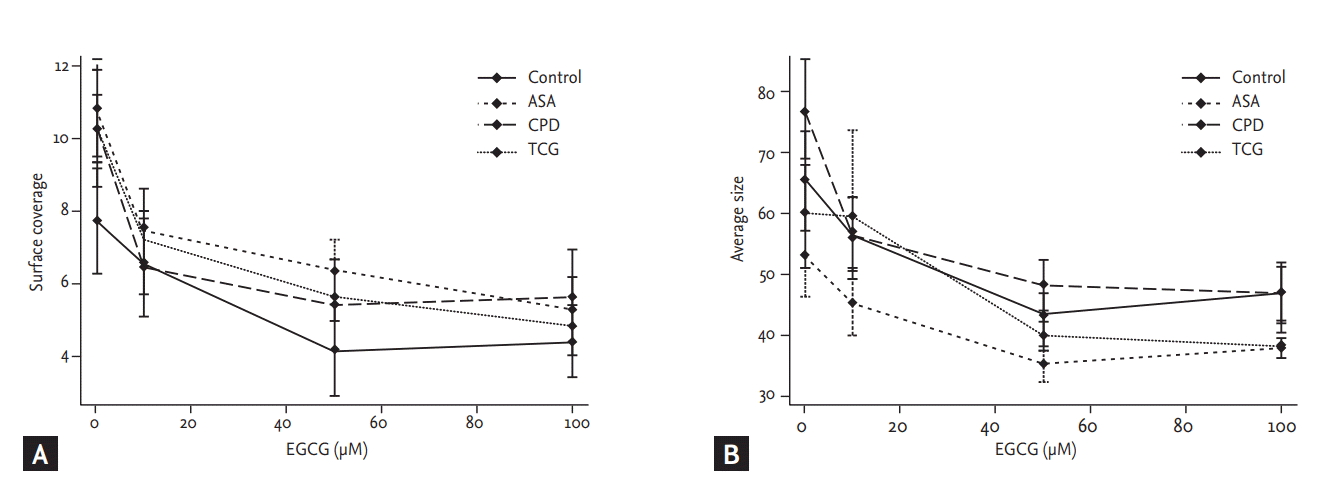
Shear stress-induced platelet adhesion
Shear stress-induced platelet adhesion (SC and AS) among four groups (SC, p = 0.45; AS, p = 0.15) revealed no significant differences among four groups (Fig. 2). However, after the treatment of EGCG significantly reduced SC in the ASA and the TCG groups (ASA, p = 0.01; TCG, p < 0.01). EGCG also significantly reduced AS in the Control, ASA, CPD groups (Control, p = 0.04; ASA, p= 0.04; CPD, p < 0.01). There were no significant differences in shear stress-induced platelet adhesion among four groups under the maximal EGCG treatment (200 ╬╝moL/L; SC, p = 0.88; AS, p = 0.51).
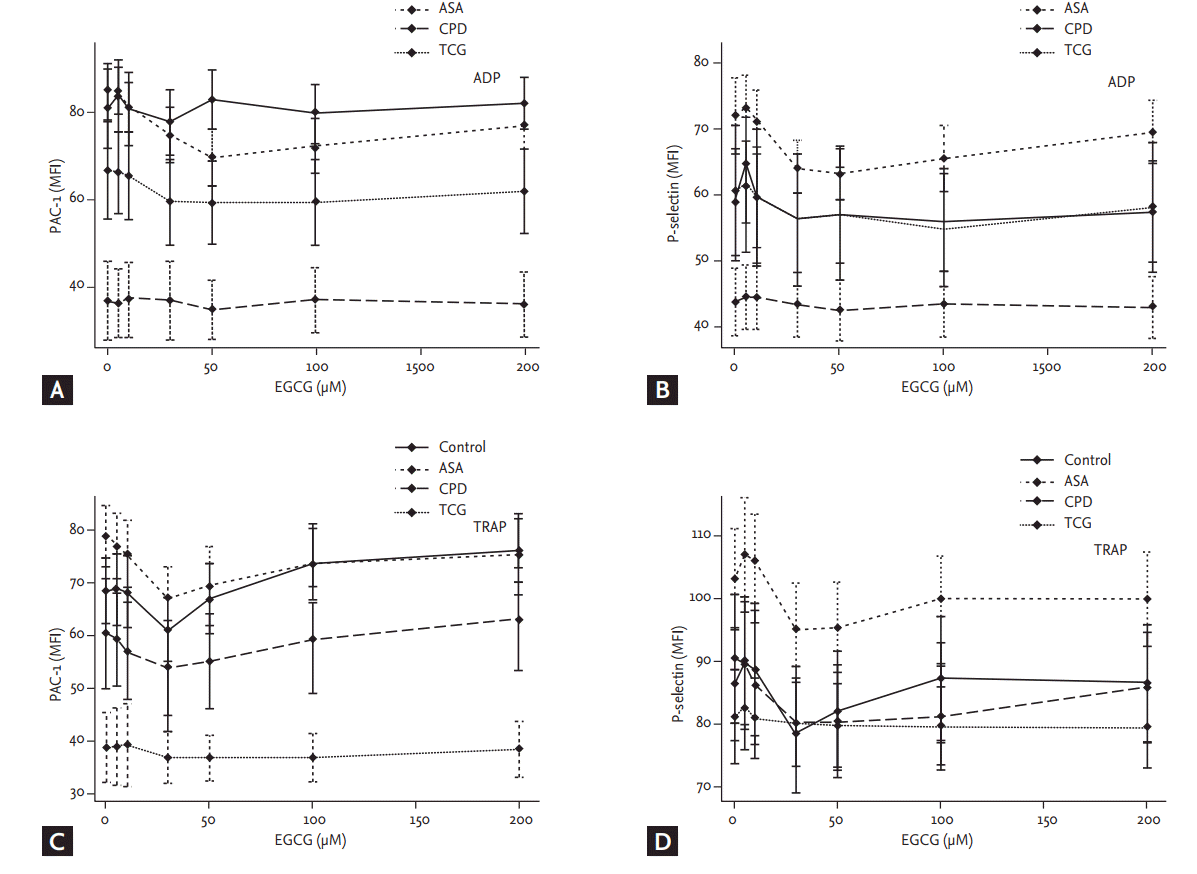
P-selectin and PAC-1 expression
Although there were no significant differences in the baseline ADP- and TRAP-induced P-selectin expressions among the four groups, the baseline ADP- and TRAP-induced PAC-1 expressions were significantly lower in the TCG group compared to the other groups (ADP, p < 0.01; TRAP, p < 0.01) (Fig. 3). EGCG had no significant effect on PAC-1 and P-selectin expressions among four groups. Finally, the ADP and TRAP-induced PAC-1 expressions were significantly lower in the TCG group compared to the other groups under the maximal EGCG treatment (200 ╬╝moL/L; ADP, p < 0.01; TRAP, p < 0.01).
DISCUSSION
The novelty of the present study is to explore the additional anti-platelet effects of EGCG in patients taking anti-platelet agents (aspirin, clopidogrel, and ticagrelor). We examined four different stimulants (ADP, TRAP, COL, and AA) induced platelet aggregations and shear stress induced platelet adhesions. Moreover, we also investigated the expression of platelet activation markers (PAC-1 and P-selectin), which were triggered by ADP and TRAP. We found that (1) EGCG strongly inhibited platelet aggregation and adhesions regardless of stimulant types, and (2) EGCG had no significant effect on platelet activation.
EGCG, a flavonoid component of green tea, has been reported to have anti-proliferative, anti-apoptotic, anti-inflammatory, and even anti-oxidant effects on the various cells in vitro [17]. Sagesaka-Mitane et al. [18] first reported anti-platelet effect of EGCG as a green tea extract. However, it was reported that EGCG had no significant effects on coagulation parameters (activated partial thromboplastin time, prothrombin time, and thrombin time) in human plasma [19].
Various agonists can activate platelets and induce their aggregation [20]. Signaling process of platelet activation and aggregation can be classified into three steps: (1) agonist-dependent early platelet activation, (2) the intermediate common signaling events, and (3) integrin-mediated final signaling pathway [21]. This is a dynamic process with intracellular and intercellular multiple feedback loops and crosstalks. Endogenous secretome from the activated platelets also exaggerates this thrombotic process. Among them, anti-platelet mechanisms of EGCG have been focused on the intracellular common signaling pathways. These included (1) inhibition of platelet cytoplasmic calcium increase [19,22], (2) increase of Ca2+-antagonistic intracellular adenosine 3ŌĆÖ,5ŌĆÖ-cyclic monophosphate (cAMP) through adenylcyclase activation and the subsequent vasodilator-stimulated phosphoprotein (VASP)-Ser157 phosphorylation [23], (3) inhibition of p38 mitogen-activated protein kinase, extracellular signal-regulated kinases-1/2, and focal adhesion kinase phosphorylation and stimulation of Syk and SLP-76 phosphorylation [24], (4) inhibition of thrombin proteolytic activity [12], and also associated with (5) cyclooxygenase-1/thromboxane A2 pathway [25,26]. Recently, Iida et al. [27] reported the effect of EGCG on human platelet activated by various stimulants including ADP, COL, ristocetin, and TRAP. EGCG only inhibited ADP-induced platelet aggregation, and failed to further suppress platelet aggregation induced by other stimulants (COL, ristocetin, and TRAP). However, in the present study, EGCG significantly reduced ADP- and COL-induced platelet aggregation in the Control group (43% inhibition for ADP, 38% inhibition for COL) (Fig. 1). However, TRAP- and AA-induced platelet aggregations were also suppressed by EGCG without a statistical significance. In addition, in the ASA group, EGCG significantly inhibited platelet aggregation stimulated by ADP-, TRAP-, and COL but not AA. Interestingly, EGCG failed to suppress all four different stimulants-induced platelet aggregation in the CPD and the TCG groups (the participants took P2Y12 receptor inhibitor). Although ADP is one of the platelet activating agonists, it also plays a role as an important cofactor for platelet activation by other stimulators [28]. Therefore, these findings suggest that EGCG inhibited platelet aggregation through the common intracellular signaling pathway (the 2nd or final step) rather than by interruption of ligand-specific platelet activation (the 1st step). In addition, no further suppressions of platelet aggregation stimulated by AA in the ASA group, and by ADP in the CPD and the TCG groups suggested no further increase of bleeding risk by EGCG in the participants who were already taking other anti-platelet agents.
Thrombus formation is also associated with shear stress [29]. Shear stress can potentiate the effects of agonists for platelet aggregation. One of novel findings in the present study was that EGCG reduced shear stress-induced platelet adhesion. This was the first report on the effect of EGCG on shear stress-induced platelet adhesion, and the action mechanism of EGCG involves most likely the second or final step of platelet aggregation. P-selectin is a membrane component of the alpha granule of platelet and its surface expression implies degranulation and platelet activation, and PAC-1 is a monoclonal antibody which binds only to the stimulated glycoprotein IIb/IIIa complex on the activated platelet [30]. Both markers are most widely used for measuring platelet activation. Previously, EGCG reduced the secretion of various factors stored in the specific granules of the activated platelets [27]. Thus, we assumed that EGCG might suppress platelet activation and decrease P-selectin and PAC-1 expressions. However, in the present study, EGCG had no significant effect on both P-selectin and PAC-1 expressions. Only baseline PAC-1 expression was significantly lower in the TCG group. Matsumura et al. [13] previously reported the similar results that EGCG failed to lower P-selectin expression although EGCG significantly inhibited platelet aggregation. Therefore, we suggest that the anti-platelet effect of EGCG might not be related to the inhibition of platelet activation.
There are several issues regarding the clinical implications of EGCG. First, the conventional anti-platelet agents (aspirin, clopidogrel, and ticagrelor) inhibit platelet aggregation dependent to their specific stimuli (ligand), and have very week anti-platelet effects on other stimuli. These ligand-specific anti-platelet effects of aspirin and clopidogrel might contribute to the aspirin or clopidogrel resistance in real world clinical practice [31]. Our findings demonstrated that EGCG inhibited platelet aggregation induced by 4 different stimuli (ADP, p = 0.04; COL, p < 0.01; TRAP, p = 0.10; AA, p = 0.10) (Fig. 1), suggesting EGCG might overcome limitations of the conventional anti-platelet agents. Second, anti-platelet effects are accompanied by the increased bleeding risk [32]. Some patient populations with high ischemic risk need dual anti-platelets or the combination therapy with anti-coagulant despite of high bleeding risk. Moreover, newer anti-platelet agents (prasugrel and ticagrelor) have been reported to be associated with increased bleed risk compared to clopidogrel. The present study revealed no further inhibition of platelet aggregation stimulated by AA in the ASA group, and by ADP in the CPD and the TCG groups. Our findings suggested that EGCG could be safe when combined with other anti-platelet agents. Thus, considering that green tea and black tee contain a considerable amount of EGCG [33], drinking green tea or black tee could provide complementary anti-platelet effect (probably with minimal bleeding risk) even in patients who already take the conventional anti-platelet agents. Furthermore, EGCG could play a role as a prototype compound for the development of novel anti-platelet agent in the future.
There are also several limitations in the present study. First, this study is not an in vivo study of taking EGCG in the participants. Previously, EGCG has shown promising results as a lead compound of a new class antiplatelet agent. However, EGCG demonstrated a low drug bioavailability [34-36]. (1) EGCG showed a low absorption profile after per os administration. (2) EGCG has relatively low distribution volume. (3) The half-life of EGCG is approximately 3 hours. Thus, EGCG should be modified to enhance its bioavailability and the further in vivo application. Before the further process, we had planned the present study to investigate the anti-platelet effect of EGCG ex vivo from the subjects who have been taking conventional anti-platelet medications. Second, relatively small sample size abated the statistical power to validate the antiplatelet effect of EGCG. EGCG significantly inhibit ADP and COL-induced platelet aggregation. TRAP and AA-induced platelet aggregation showed the marginal, not statistically significant, suppression by EGCG. Third, the detailed mechanisms on anti-platelet effect of EGCG were not studied. Despite of these limitations, the major strength of the present study is that the data were derived from human samples especially in the subjects already taking conventional anti-platelet agents. The present findings might provide fundamental data for newer anti-platelet drug development from EGCG derivatives.
In conclusion, EGCG significantly inhibited the human platelet aggregation without any changes on P-selectin and PAC-1 expressions. However, AA-induced platelet aggregation in the participants treated with aspirin, and ADP-induced platelet aggregation in the participants treated with P2Y12 receptor inhibitors (clopidogrel or ticagrelor) were not suppressed additionally by EGCG, suggesting that EGCG might have a potential role as a new class anti-platelet agent without increasing bleeding complications.
KEY MESSAGE
1. Epigallocatechin-3-gallate (EGCG) significantly reduced adenosine diphosphate and collagen-induced platelet aggregation in dose-dependent manner.
2. EGCG also suppressed shear stress-induced platelet adhesion, but had no effect on P-selectin and PAC-1 expressions.
3. EGCG did not suppress platelet aggregation any further in subjects already took anti-platelet agents. It implied no increased bleeding risk by EGCG.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





