INTRODUCTION
Vanherweghem et al. [
1] were the first authors to observe, in an epidemiological study in Belgium, that Chinese herb nephropathy (CHN) can be induced by aristolochic acid (AA), one of the major constituents of
Aristolochia fangchi [
1-
4]. The material analyzed in the Belgian cases was a mixture of aristolochic acid I (AA-I) and aristolochic acid II (AA-II) [
3,
5,
6]. Thereafter, many investigators in other studies were able to induce CHN in a variety of experimental animals using an AA mixture [
6]. Debelle et al. [
6,
7] succeeded in inducing CHN in salt-depleted Wistar rats, and demonstrated that the AA mixture was the causative substance. Therefore, the designation of CHN was changed to the current term of aristolochic acid nephropathy (AAN). Sato et al. [
8] demonstrated that AA-I was more toxic than AA-II and Xue et al. [
9] established an animal experimental model of acute AAN through an intra-abdominal injection of AA-I in C57BL/6 rats.
In most of the European CHN cases, rapidly progressive interstitial nephritis progressed to end-stage renal failure requiring renal replacement therapy [
1,
6,
10,
11]. In Asian countries, however, most cases were accompanied by subacute interstitial nephritis and acquired Fanconi syndrome (aFS) without progressing to ERSD. These cases were managed by correcting the electrolyte imbalance and through conservative treatments [
12-
15]. The hypokalemia observed in aFS may cause renal injury because previous animal experiments have demonstrated the presence of hypokalemic nephropathy (HKN) with tubulointerstitial nephritis in renal medullas [
16-
19]. Its pathophysiology remains uncertain [
20].
Given the aforementioned background information, we aimed to induce aFS in an animal model of acute AAN with pure AA-I, rather than an AA mixture. In addition, this study also aimed to investigate the role of hypokalemia in acute AAN, as well as to identify the pathogenesis of AAN.
METHODS
Animal protocols
The current animal experiment was approved by the Institutional Review Board of College of Medicine, The Catholic University of Korea. The experimental rats consisted of 40 male C67BL/6 mice 8 weeks in age (Orient Bio, Seongnam, Korea), weighing approximately 20g (
Table 1). They were bred in an animal laboratory at the Catholic University at 24┬░C with 12-hour increments of alternating light and dark under controlled humidity.
The experimental animals were housed 5 to a cage and fed a normal potassium (K) diet (1% K) and primary distilled water for a week. They were then divided into the following four groups.
(1) Group I (n = 10), served as the control group, given a normal K diet.
(2) Group II (n = 10) given a low K diet (0.01% K, Research Diet, New Brunswick, NJ, USA) to induce hypokalemia.
(3) Group III (n = 10) fed a normal K diet following a single intra-abdominal administration of AA-I at 10 mg/kg (Sigma Chemical Co., St. Louis, MO, USA).
(4) Group IV (n = 10) fed a low K diet with the same AA-I administration as group III.
The AA-I (50 mg, AA-I > 95%, Sigma Chemical Co.) that was used in the current study was dissolved in 50 mL of warm saline and then diluted to 1 mg/mL. At the beginning of the experiment, 10 mg/kg of AA-I was prepared and intra-abdominally administered in 0.2 mL saline as a single dose. To induce hypokalemia, the rats were given a low K diet and primary distilled water.
The body weights of the rats were measured at 2 weeks (
Table 1). Five rats in each group were then anesthetized with 16.5% urethane(10 mL/kg) and their abdominal aortae were punctured with a heparin-filled syringe. Blood samples were collected and the rats were sacrificed. The renal tissue was perfused, fixed, and extracte d through the heart. Two days prior to this procedure, the five remaining rats in each group were placed in a metabolic cage. After a 1-day accommodation period, the remaining rats in each group were subjected to a 24-hour urine collection protocol.
Blood and urine tests
To measure the serum concentrations of sodium (Na), K, blood urea nitrogen (BUN), creatinine, and total carbon dioxide (TCO2), we collected approximately 0.2 to 0.3 mL of whole blood through the abdominal vena cava from each rat. Subsequently, using an i-STAT EC8+06F04-01 cartridge (Abbott Laboratories, Princeton, NJ, USA), the blood samples were placed in an i-STAT analyzer (AbbottLaboratories) and analyzed.
Immediately after collecting urine samples, the volume and pH were measured. To measure the urine concentrations of Na, K, blood glucose, protein, and creatinine, we collected urine samples over the course of 24 hours and froze them at ŌĆō20┬░C. Urine Na and K were quantified using ion selective electrodes (Bayer, Pittsburgh, PA, USA). Creatinine was quantified with a Creatinine Reagent kit (Bayer) based on the Jaffe reaction. In addition, glucose was quantified with an ADVIA 1650 (Bayer, Tokyo, Japan) using a Glucose Hexokinase kit.
Histopathological examination
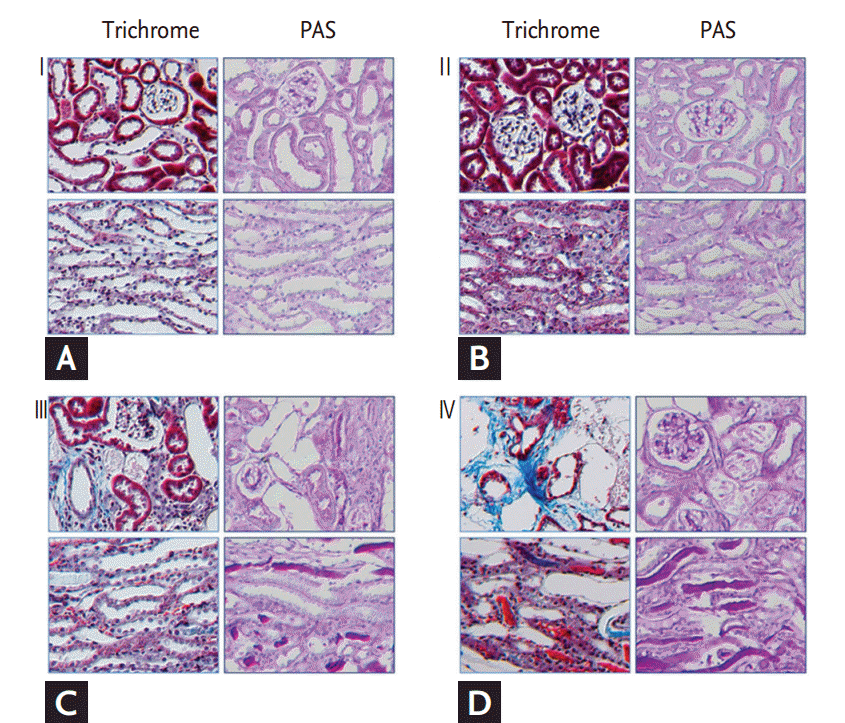
After the mice were anesthetized with 16.5% urethane (10 mL/kg body weight) for immunohistochemistry, the whole blood was washed with 0.01 M phosphate-buffered saline (PBS, pH7.4) through cardiac puncture. Perfusion and fixation were performed with 2% paraformaldehyde- lysine-periodate for 10 minutes, and then the kidneys were promptly collected. The kidneys were sectioned at a thickness of 2 mm that included the renal papilla and fixed in the same fixative mentioned above at 4┬║C for 6 to 12 hours. After dehydration, the tissue samples were embedded with polyester wax (Polyscience, Warrington, PA, USA). The 4 ╬╝m section slides were prepared and covered with gelatin in order to perform immunohistochemistry analysis. For the histopathologic examinations, we performed hematoxylin and eosin (H&E) staining, periodic acid-Schiff (PAS) staining, and MassonŌĆÖs trichrome (Trichrome) staining.
Renal histopathology
In each group, all of the samples of kidney tissue that were stained with H&E, PAS, and Trichrome were analyzed with a light microscope (Olympus BX51, Olympus, Tokyo, Japan) at a magnification of ├Ś200. Based on a modified scoring system [
21], the results were scored as follows:
(1) Tubular necrosis: 0, normal; 1, a single necrotized tubule; 2, multiple necrotized tubules; 3, a cluster of tubular necrosis.
(2) Tubular atrophy: 0, normal; 1, a single necrotized tubule; 2, atrophy of multiple tubules; 3, a cluster of tubular necrosis.
(3) Lymphocyte infiltration: 0, none; 1, diffuse; 2, a cluster of lymphocyte infiltration; 3, overall infiltration of lymphocytes.
(4) Interstitial fibrosis: 0, none; 1, mild fibrosis; 2, moderate fibrosis; 3, severe fibrosis.
(5) Tubular enlargement: 0, none; 1, mild enlargement; 2, moderate enlargement; 3, severe enlargement.
(6) Amount of cast within the tubule: 0, none; 1, mild; 2, moderate; 3, severe.
Immunohistochemistry
For immunohistochemistry and Western blot analysis, ╬▓-actin (Sigma Inc.), ╬▒-smooth muscle actin (╬▒-SMA, Sigma Inc.), E-cadherin (BD Transduction Lab., San Diego, CA, USA), and F4/80 (Serotec Inc., Raleigh, NC, USA) were used.
The wax of the 4 ╬╝m sectioned tissues was removed through alcohol and xylene, and the sections were then immersed in 0.4% methanolic H2O2 for 30 minutes, followed by 0.05% Triton X for 15 minutes. After incubation in 2% donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 hour, these sections were immersed in primary antibodies, ╬▒-SMA 1:1,000, E-cadherin 1:200, and F4/80 1:50 overnight at 4┬║C.
The following day, the sections were washed with 0.01 M PBS and then immersed in donkey-anti-rat or donkey-anti-mouse (Jackson ImmunoResearch Laboratories) secondary antibodies diluted to 1:200 and incubated for 2 hours at room temperature. Immunoreactivity was detected by incubation in Tris buffer (pH 7.6) containing 0.05% 3,3ŌĆ▓-diaminobenzidine-4HCl and 0.005% H2O2. These sections were then counterstained with MeyerŌĆÖs hematoxylin and mounted with Canada balsam for examination.
Computer-assisted microscopy evaluation of F4/80
After F4/80 immunohistochemistry, the slides were divided into renal cortex and medulla groups. At a magnification of ├Ś400, 5 to 10 regions were randomly selected for photography and images were analyzed with Image-Pro Analyzer 6.3 version 6.3.0.512 for Windows XP/Vista (Media Cybernetics Inc., Rockville, MD, USA).
Western blot analysis
For the quantitative analysis of proteins, we performed Western blot analysis. The experimental animals were anesthetized with 16.5% urethane (10 mL/kg) and then perfused with 0.01 M PBS through the heart. After the blood was rinsed, the kidneys in each of the animals were extracted, divided into the cortex and medulla, mixed with 80┬║C hot lysis buffer (1% sodium dodecyl sulfate, 1 mM Na3VO4, 10 mM Tris, pH 7.5), and homogenized. After centrifugation, protein was extracted and quantified using the bicinchoninic acid assay (Pierce, Thermo Scientific, Waltham, MA, USA). Following electrophoresis, the proteins were transferred to a nitrocellulose membrane and incubated for 1-hour with phosphate buffered saline Tween (PSBT; PBS containing 0.1% Tween 20) containing 5% skim milk. The membranes were then stained with primary antibodies (╬▓-actin 1:300,000, E-cadherin 1:1,000, and ╬▒-SMA 1:5,000). The following day, the membranes were rinsed with PBST and donkey-anti-rat or donkey-anti-mouse (Jackson ImmunoResearch Laboratories) secondary antibodies were applied at a 1:2,000 dilution and incubated at room temperature for 2 hours. Color was developed by enhanced chemiluminescence and exposure to light on X-ray film. Following this step, the bands were analyzed with Multi Gauge (Fujifilm Corp., Tokyo, Japan).
Statistical analysis
All data are expressed as mean ┬▒ SD. The control and experimental values were compared with the Mann-Whitney U test and Kruskal-Wallis test. We also analyzed parametric variables using independent sample t tests and one-way analysis of variance. Statistical analysis was performed with SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). A p < 0.05 was considered statistically significant.
DISCUSSION
The acute AAN mouse model in this study was induced by intra-abdominal injection of a single 10 mg/kg dose of AA-I based on previous animal studies which suggested that nephrotoxicity not only occurred following the administration of AA-I at a dose of 10 to 20 mg/kg in C57BL/6 rats, but also that the LD50 level of AA-I was 37 mg/kg [
8,
9]. A comparison between the experimental groups and the control group in this study suggests that this dose was appropriate. In addition, the HKN mouse model was correctly induced in group II because (1) serum potassium concentrations (K
+) were 2.8 ┬▒ 0.2 and 3.3 ┬▒ 0.3 mEq/L even in group IV with the AA-I injection together with hypokalemia and (2) urine pH was higher and urine output was larger in group II than in the control group. Previous animal studies for inducing HKN have shown that serum K
+ was 3.3 ┬▒ 0.21 mEq/L [
22] with elevated urine pH and urine output levels [
17]. Our results are consistent with the true HKN animal model (
Table 2).
Serum and 24-hour urine biochemistry at 2 weeks showed that there was no significant difference in BUN between groups III and IV, but serum creatinine levels were lower in group IV. This may suggest that nephrotoxicity was lower in group IV than group III. However, since body weight declined more in group IV, nephrotoxicity was actually higher in that group, where AAN and HKN were both induced. This indicates that a small amount of muscle may be included in the decreased body weight.
FeNa and FeK levels increased in the 24-hour urine output time period. Glycosuria and proteinuria were more pronounced in groups III and IV than the other groups (
p < 0.05) (
Table 4), as were polyuria and urinary alkalization (
Table 2). These results are similar to previous animal studies of aFS [
23,
24]. Therefore, aFS appears to have been induced by the administration of AA-I (10 mg/kg) in groups III and IV despite the fact that K
+ was 3.9 ┬▒ 0.4 mEq/L in group III. Since these findings are not consistent with previous reports that hypokalemia accompanies aFS in humans, AA-I may not induce aFS. According to Sato et al. [
8] and Pozdzik et al. [
21], AA-I has the greatest toxicity among the AA variants, and severe histopathologic damage developed rapidly in the proximal renal tubule of the cortex after 5 days of administration of an AA mixture (AA-I 40% and AA-II 60%) at a dose of 10 mg/kg in Wistar rats. In light of these studies, we may have failed to detect the change in serum K
+ associated with aFS because either the toxicity of AA-I exceeded our estimates for severe renal failure due to hyperkalemia, or we collected our samples too late. The histopathologic findings showed that acute proximal tubular injury was localized to the renal cortex in groups III and IV. Therefore, AA-I may play a key role in the pathogenesis of aFS associated with AAN.
Our finding that proximal tubular injury was localized to the renal cortex was similar to that of previous animal studies of AAN using AA mixtures. Unfortunately, we failed to observe inflammatory cell infiltration, renal tubular atrophy, and interstitial fibrosis reported in those same previous studies [
5-
9,
21]. This might suggest that the severe toxic effect of AA-I (10 mg/kg) may induce apoptosis of the epithelial cells lining the proximal renal tubule without inflammation when it reaches the glomerular filtration stage. Some in vitro studies support this hypothesis. Hsin et al. [
25] studied LLC-PK1 cells (the stalk of the porcine proximal renal tubular epithelial cells) exposed to AA. As there was an abrupt increase in the concentration calcium ions which causes stress to the endoplasmic reticulum and mitochondria and activates the caspase pathway, this eventually led to apoptosis. Balachandran et al. [
26] also found that AA-I induced the activation of caspase 3/7 which leads to apoptosis in renal tubular cells. Moreover in an experiment with LLC-PK1 cells exposed to AA-I, Li et al. [
27] found that DNA damage and cell division impairment occurred prior to renal tubular necrosis or apoptosis. Therefore, we assume that AA-I may not damage the epithelial cells lining the proximal renal tubule through the infiltration of inflammatory cells or immune reactions, but rather by a direct toxic effect that induces apoptosis.
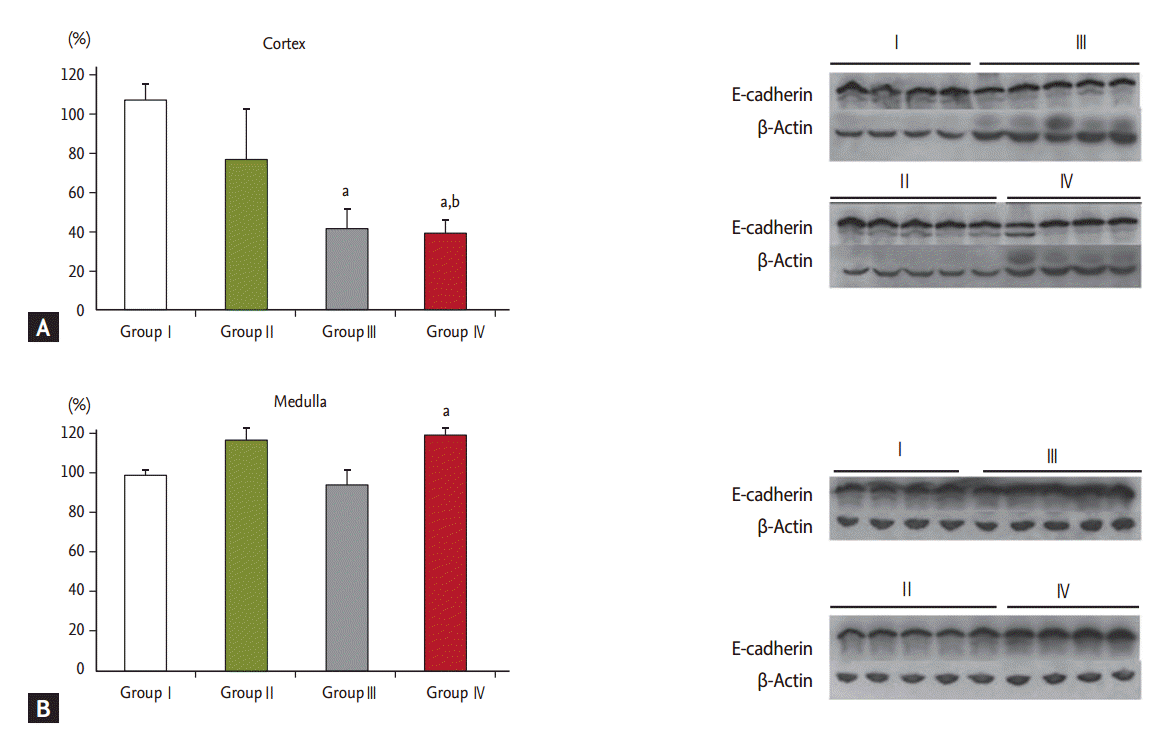
In addition, Western blot analysis of the renal cortex revealed an increase in the expression of ╬▒-SMA and F4/80 in groups III and IV and a significant decrease in E-cadherin (
Figs. 2-
4). There was no evidence of infiltration of inflammatory cells or renal fibrosis suggestive of the epithelial to mesenchymal transition (EMT) of the epithelial cells lining the proximal renal tubule. Pozdzik et al. [
21] reported that AA damaged the renal tubules by activating antioxidative enzymes and causing mitochondrial damage, and that the progressive renal tubular atrophy was associated with a lack of regeneration of the epithelial cells lining the proximal renal tubule with secondary apoptosis being due to the activation of caspase-3. Vimentin and ╬▒-SMA positive cells increased in the interstitial areas where transforming growth factor ╬▓ was expressed, suggesting that the fibroblasts between the renal tubules had proliferated and myofibroblasts had been activated, as opposed to the presence of EMT of the epithelial cells lining the proximal renal tubule as a result of the developing collagen deposits and interstitial fibrosis [
21,
28,
29]. Therefore, because of the histopathologic findings and Western blot data, we assume that pure AA-I, as opposed to the AA mixture, is directly toxic or induces apoptosis in proximal tubular cells without interstitial fibrosis. However, we failed to detect the effects of hypokalemia in the AAN animal model since there were no significant differences in the immunohistochemical and Western blot results in the renal cortex between groups III and IV.
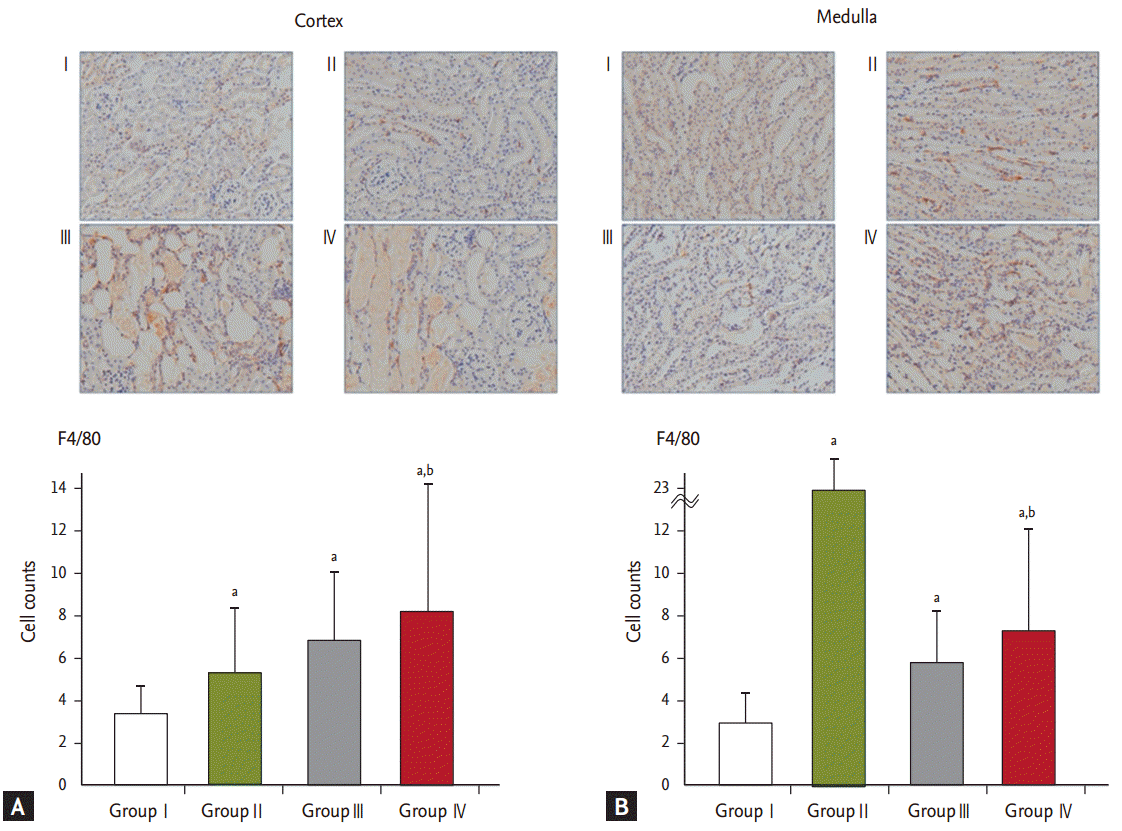
Interestingly, the expression of F4/80, detected using an F4/80 antibody for macrophages, increased in the renal cortices of groups III and IV. Wilson et al. [
30] showed that macrophages could trigger inflammatory responses and apoptosis in the interstitial areas of the renal tubule, and therefore may be associated with renal injury and regeneration. Moreover, in an animal experimental model of cyclosporine-induced renal injury, macrophages induced immunological or non-immunological damage leading to apoptosis, as reported by Ghee et al. [
31]. From this finding, it could be inferred that macrophages may also play a role in the connection with apoptosis or cell regeneration in this study employing pure AA-I.
Our analysis of changes in the renal medulla showed that the expression of F4/80 increased more in group II than in the other groups (
p < 0.001). While it has been postulated that hypokalemia in HKN induces the contraction of renal arterioles as a result of ischemia arising from decreased perfusion in the renal medulla [
32-
34], an animal study of the remnant kidney by Kang et al. [
35] noted that macrophages infiltrated the renal cortex and medulla. This resulted in the disappearance of capillary vessels and the release of cytokines from macrophages that suppressed the expression of vascular endothelial growth factor (VEGF) in the epithelial cells lining the renal tubules [
35]. In addition, another study of an HKN model by Reungjui et al. [
22] documented that the expression of macrophage monocyte chemoattractant protein-1 and tumor necrosis factor ╬▒ increased in the renal medulla while the production of endothelial nitric oxide and VEGF decreased. We therefore speculate that macrophages play a key role in the pathophysiology of HKN, as shown in the case of AAN.
However, this current study has limitations. The first is that we failed to induce interstitial nephritis through the administration of AA-I. In addition, there were almost no findings suggestive of renal tubular atrophy, interstitial fibrosis, or infiltration of inflammatory cells, all of which have been described in previous animal studies of AAN. Instead, very severe acute renal tubular necrosis was confined to the renal cortex. These acute changes may have made it difficult to observe the effects of hypokalemia and to generate true aFS in this AAN animal model using AA-I, rather than an AA mixture. In addition, because all the results were obtained after 2 weeks, our study may have failed to detect an effect of AA-I during the first days of exposure or in the acute stage. For these reasons, to establish when AA-I induces apoptosis of proximal renal tubule cells or has a specific toxic effect on them will require further studies using a lower dose of AA-I, an examination of earlier changes, and a measurement of capase-3 and cytochrome c. The second limitation was that a Western blot analysis of F4/80 was not performed to obtain objective results. Therefore, our suggestion about the role of macrophages is only minimally supported by our findings. A third limitation is that we may have failed to investigate significant differences in the renal cortex and medulla between groups III and IV, because group IV was simultaneously induced by both hypokalemia and pure AAI. Further investigation is needed on AA-I induction of AAN in an established HKN animal model.
We conclude that AA-I, one of the AA variants, plays a key role in the pathogenesis of aFS in the early stages of AAN, and has a powerful toxic effect on the epithelial cells lining the proximal renal tubule by non-immunological mechanisms. We also suggest that macrophages play a key role in the pathogenesis of both AAN and HKN. However, due to the limitations of the present study, we did not detect an effect of hypokalemia.




 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





