


INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm characterized by the clonal expansion of hematopoietic stem cells with a reciprocal translocation between chromosomes 9 and 22. This Bcr-Abl fusion gene is transcribed into a chimeric Bcr-Abl mRNA, resulting in activation of tyrosine kinase and down-stream kinases that prevent apoptosis [1]. Until the development of Bcr-Abl tyrosine kinase inhibitors (TKIs), patients with CML such as patients with acute leukemia, had poor prognosis, and the only treatment strategy for cure was stem cell transplantation. However, CML treatment has shown impressive success with the development of Bcr-Abl TKIs. Imatinib mesylate was the first Bcr-Abl TKI to be developed for the treatment of CML [2-4]. In long-term follow up data from the IRIS (International Randomized Study of Interferon and STI571) trial, the major molecular response rate and overall survival rates were 86% and 85%, respectively, after 8 years [5].
Despite high response rates, resistance to TKIs develops in a significant number of CML patients [6,7]. Various strategies have been investigated to overcome this resistance, including increasing the dose of imatinib, developing novel drugs that act on the mutated adenosine triphosphate (ATP)-binding sites of the Bcr-Abl fusion protein, and combining imatinib with other drugs to block the down-stream signals of the Bcr-Abl protein [7,8]. Recently, second generation TKIs (nilotinib and dasatinib) have been developed to overcome imatinib resistance [9,10]. Although these agents have shown superior outcomes to imatinib, some resistances, such as the T315I mutation, have not been overcome. Furthermore, some patients cannot be treated with a TKI alone. Those patients must be treated by stem cell transplantation, which has a high treatment-related mortality rate. For these reasons, development of new strategies for eradication of resistant CML cells is needed.
The Bcr-Abl oncogenic pathway is the main causative factor in leukemogenesis of CML. Other signaling molecules reported to be involved in the pathogenesis of CML include the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (mTOR) pathway, the RAS/mitogen-activated protein kinase pathway, the Janus-activated kinase/signal transducer and activator of transcription (JAK/STAT) pathway, the Hedgehog pathway, and the Wnt pathway [11]. Considerable research on inhibitors of these pathways is being carried out. It has been reported that iron overload may lead to proliferation of several types of cancer. Our institute previously reported the effects of iron chelator on lymphoma and leukemia cells [12,13]. Furthermore, cancer cells typically require more iron than normal cells to mediate their rapid DNA synthesis and growth [14,15]. Treatment of hepatocellular carcinoma or acute leukemic cancer cells with an iron chelator inhibited proliferation and induced apoptosis [16]. The orally administered iron chelator, deferasirox, is a widely used drug for the treatment of iron overload, which is caused by repetitive transfusion in patients with hematologic malignancies. Recently published prospective trials have revealed that deferasirox leads to hematologic improvement in patients with myelodysplastic syndrome [17]. A case report showed that deferasirox administration led to a complete remission in a patient with chemotherapy-refractory acute monocytic leukemia [18]. These anticancer effects could be explained by the down-regulation of the NF-╬║B, mTOR, and/or Wnt pathways by deferasirox [19,20]. Thus, an anti-leukemic effect of deferasirox may be expected in CML patients. Such an antileukemic effect would also be expected in imatinib-resistant CML cells by blocking another pathway down-stream of the Bcr-Abl signaling pathway [19,21,22]. The aim of this study was to identify the synergistic effects of deferasirox and imatinib and to confirm the effect of this combination treatment on imatinib-resistant cells.
METHODS
Cell lines
This study was conducted with the human CML cell lines K562 and KU812. The K562 cell line was purchased from the Korea Cell Line Bank (KCLB, Seoul, Korea), and the KU812 cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were maintained according to instructions provided by the ATCC and KCLB. Both cell lines were maintained in Roswell Park Memorial Institute Media 1640 (RPMI-1640) supplemented with 10% fetal bovine serum and antibiotic-antimycotic (100├Ś, Life Technologies, Waltham, MA, USA) at 37┬░C and 5% CO2 in a humidified incubator.
Drugs and reagents
The oral iron chelator deferasirox was provided by Novartis (Basel, Switzerland), and imatinib mesylate was purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA; anti-BCR-ABL, anti-pBcr-Abl, anti-Akt, anti-caspase 3/9, anti-poly [ADP-ribose] polymerase [PARP], anti-STAT3, anti-pSTAT3, anti-pIkBa, and anti-poly-glycogen synthase kinase 3 [pGSK3] ╬▒/╬▓), BD Biosciences (Franklin Lakes, NJ, USA; anti-Bax and anti-╬▓-actin), Santa Cruz (Santa Cruz, CA, USA; anti-IkBa), and Sigma (anti-NF-╬║B).
Cell viability assay
Cell viability was evaluated by a colorimetric assay (Ez-cytox Kit, High Sensitive Water Soluble Tetrazolium). K562 and KU812 CML cells were seeded in 96-well plates (1.5 ├Ś 104 cells/well) and were treated with various concentrations of deferasirox (10, 20, 50, and 100 ╬╝M) or imatinib (0.5, 1, 5, 10, 20, 50, and 100 ╬╝M). After 24, 48, or 72 hours of incubation, 10 ╬╝L per well of the Kit reagent was added to each well. The cells were then incubated at 37┬░C for an additional 4 hours. The absorbance of the treated cells at 450 nm was measured by using a microtiter plate reader. The combined effects of deferasirox and imatinib (D/I: 10/1, 10/5, 10/10, 20/1, 20/5, or 20/10 ╬╝M) were evaluated by the same method. The synergistic effect of deferasirox and imatinib was determined by calculating the combination index (CI) by using Calcusyn Software (Biosoft, Ferguson, MO, USA) [23,24]. A value of CI < 1 indicates synergism, CI = 1 indicates an additive effect, and CI > 1 reflects an antagonist interaction.
Cell cycle analysis
K562 and KU812 cells were plated in 6-well plates with RPMI media and treated with deferasirox (10, 20, 50, and 100 ╬╝M) or imatinib (0.5, 1, 5, 10, 20, 50, and 100 ╬╝M) alone or in combination (D/I: 10/1, 10/5, 10/10, 20/1, 20/5, or 20/10 ╬╝M). After treatment, cells were harvested at designated times and washed twice with phosphate buffered saline (PBS). Cells were fixed in PBS containing 5 mM ethylenediaminetetraacetic acid and 85% ethanol for 30 minutes at 4┬░C. The cells were then treated with 20 ╬╝g/mL RNase A and 50 ╬╝g/mL propidium iodide (Sigma) for 30 minutes and analyzed with a Cytomix FC 500 (Beckman Coulter, Brea, CA, USA) flow cytometer. Ten thousand events were counted per sample. The percentages of the cell population at the G0-G1 and G1 phases were calculated.
Apoptosis analysis
CML cells treated with deferasirox (10, 20, 50, and 100 ╬╝M) or imatinib (0.5, 1, 5, 10, 20, 50, and 100 ╬╝M) alone or in combination (D/I: 10/1, 10/5, 10/10, 20/1, 20/5, or 20/10 ╬╝M) were evaluated by annexin V/propidium iodide staining and flow cytometry.
Western blot analysis
To evaluate the mechanisms of apoptosis after treatment of CML cells with deferasirox and imatinib, we performed western blot analyses. Whole cell extraction was conducted by using RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate, and 1% sodium deoxycholate pH 7.4) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 ┬Ąg/mL peptasin A, 10 ┬Ąg/mL aprotinin, and 5 ┬Ąg/mL leupeptin). The protein concentrations were measured by using Bio-Rad protein assay kits (Bio-Rad, Hercules, CA, USA). The protein lysates were resolved by sodium docdecyl sulfate polyacrylamide gel electrophoresis, then transferred onto nitrocellulose membranes (HybondTM-P, Amersham Biosciences Co., Piscataway, NJ, USA), blocked with PBS containing 0.2% Tween 20 and 5% non-fat dry milk, and incubated with primary antibody. The primary antibodies against the following proteins: Bcr-Abl, pBcr-Abl, Bax, caspase 3, caspase 9, PARP, STAT3, pSTAT3, IkB╬▒, pIkB╬▒ and NF-kB, pGSK3 ╬▒ / ╬▓, ╬▓-catenin, and ╬▓-actin.
Imatinib-resistant cell line
Imatinib-resistant CML cell lines (K562R, KU812R) were induced by continuous exposure to imatinib in stepwise increments from 0.5 nM to 1 ╬╝M after every 10 days of culture. Viable imatinib-resistant CML cells were maintained in 1 ╬╝M imatinib. To confirm the resistance of the cells, imatinib-sensitive cells (K562 and KU812) and imatinib-resistant cells (K562R and KU812R) were compared by using the cell viability assay and Western blot analysis. To determine the effect of co-treatment in imatinib-resistant cells, the induction of apoptosis and the expression level of Bcr-Abl protein and down-strea m signaling molecules were evaluated by western blot.
RESULTS
Co-treatment with deferasirox and imatinib inhibits proliferation of human CML cells
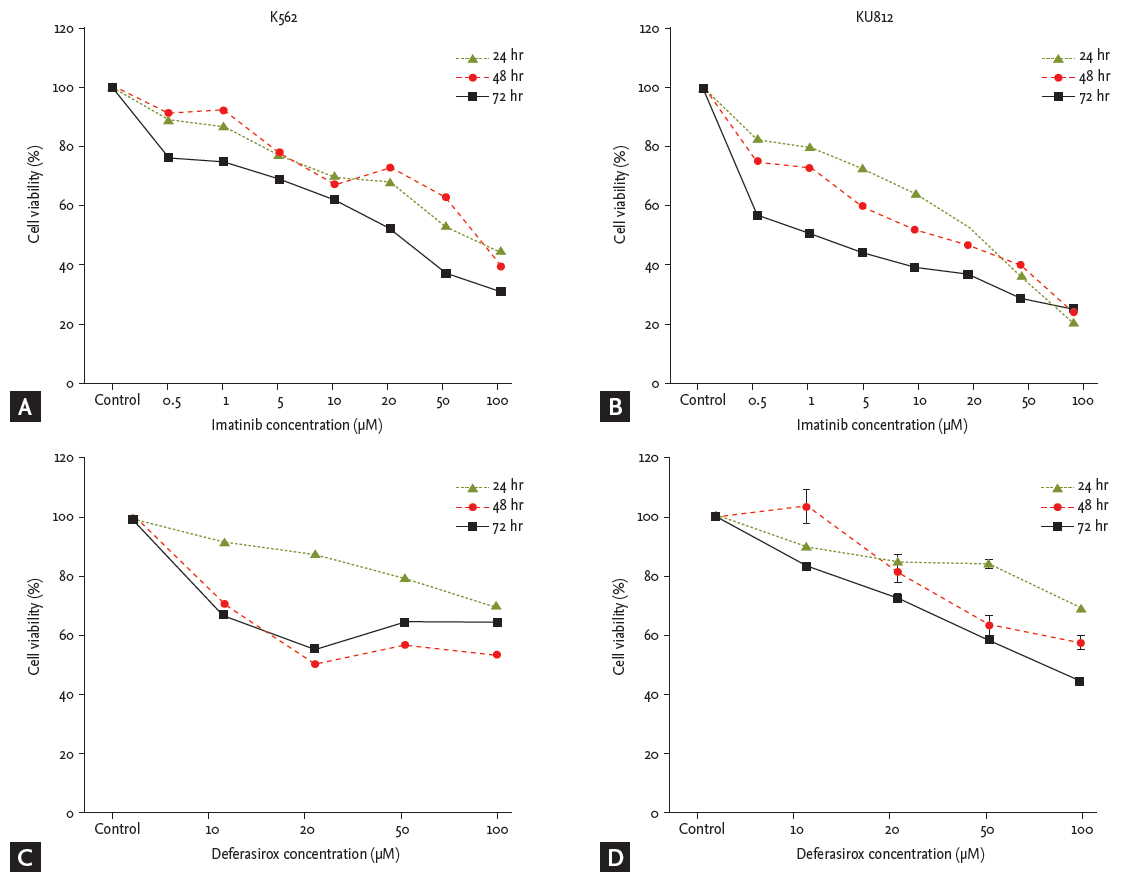
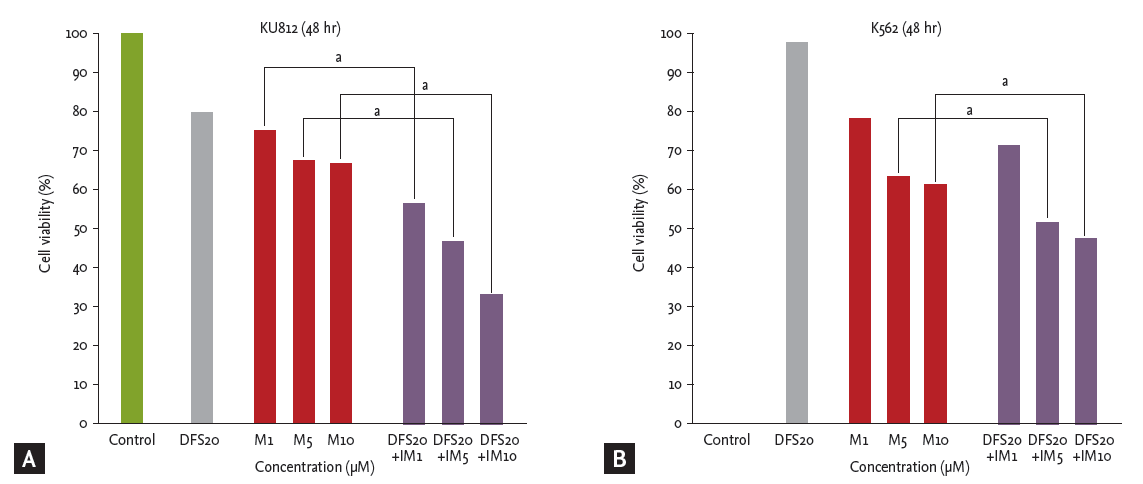
Imatinib and deferasirox both produced a dose- and time-dependent reduction of cell viability of both K562 and KU812 cells (Fig. 1). After co-treatment of KU812 cells with deferasirox 20 ╬╝M and imatinib 10 ╬╝M, cell viability was reduced to 33.7%, whereas treatment with deferasirox 20 ╬╝M or imatinib 10 ╬╝M alone reduced the cell viability to only 80.2% and 66.5%, respectively (Fig. 2A). This trend was similar for K562 cells (Fig. 2B). To determine whether there was a synergistic effect of the two drugs, we analyzed the results by the Chou and Talalay [23] method, which is the most widely used method to determine the effects of drug combinations. The CI values for deferasirox and imatinib at all of the combination concentrations were all less than 1 (CI, 0.18 to 0.67). This shows that co-treatment with these two drugs produces synergism.
Co-treatment with deferasirox and imatinib induces cell cycle arrest at the subG1 phase
To determine the effects of deferasirox and imatinib on cell cycle progression, both CML cell lines were treated with each drug and analyzed by flow cytometry. Treatment with deferasirox or imatinib singly induced an accumulation of cells in the subG1 phase for both the cell lines (Fig. 3). Co-treatment with both drugs led to a greater cell accumulation in the subG1 phase than single treatment. Cells in S phase were decreased.
Co-treatment with deferasirox and imatinib induces apoptosis and significantly down-regulates the Bcr-Abl down-stream pathway of human CML cells
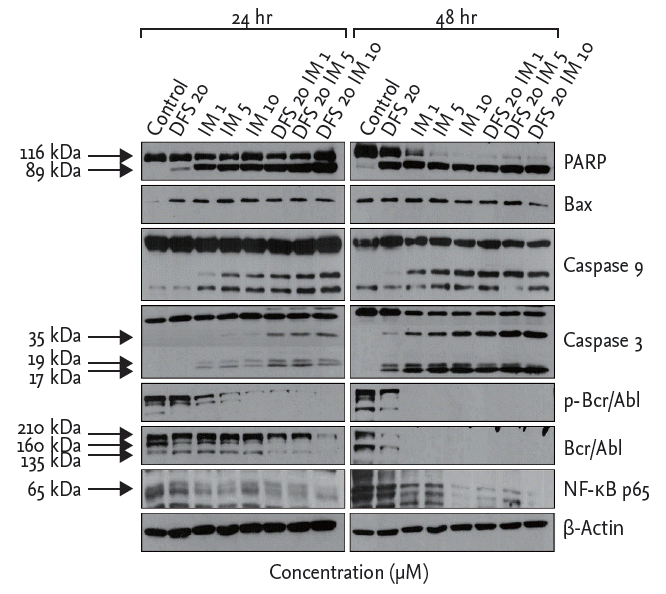
To determine the effects of deferasirox on apoptosis and protein expression after treatment with deferasirox or imatinib, singly or in combination, we measured the expression levels of proteins by using Western blot analysis. Compared to single treatment, the results of co-treatment with deferasirox and imatinib showed increased expression of cleaved PARP and cleaved caspase 3/9 as markers of apoptosis. Expressions of Bcr-Abl, pBcr-Abl, and NF-╬║B of K562 cells were decreased after the combination treatment (Fig. 4). Similar results were obtained with KU812 cells.
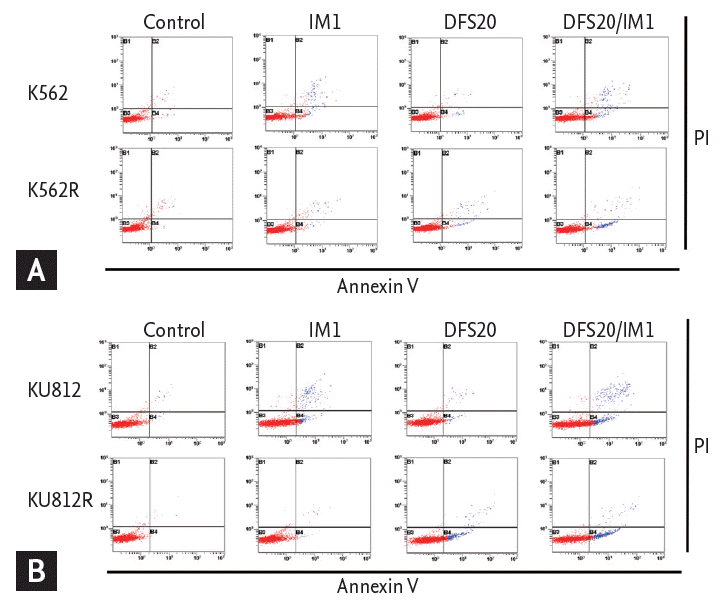
Co-treatment with deferasirox and imatinib induced apoptosis in imatinib-resistant CML cells
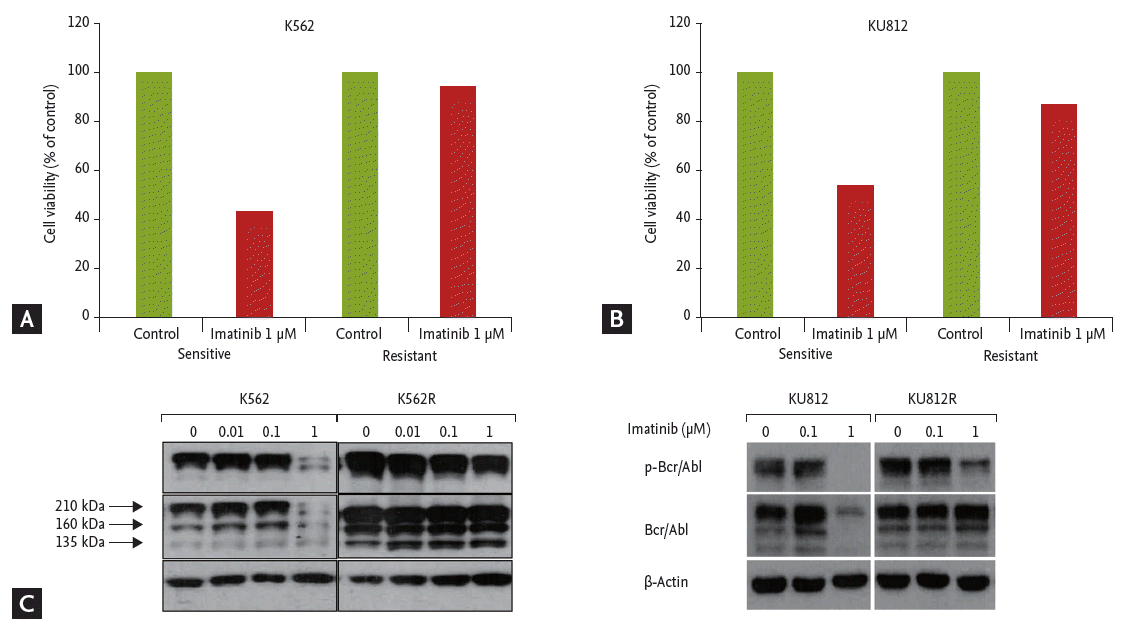
Imatinib-resistant CML cell lines were produced by repeated exposure to gradually increasing concentrations of imatinib, from 0.01 to 1 ╬╝M. After treatment with imatinib, cell viabilities were compared between imatinib-sensitive and resistant cell lines by colorimetric assay. Only 43% of K562 cells survived, but 95% of imatinib-resistant K562 (K562R) cells survived after treatment with 1 ╬╝M imatinib (Fig. 5A). For KU812 cells, 52% survived after 1 ╬╝M of imatinib, but 90% of imatinib-resistant KU812 (KU812R) cells survived (Fig. 5B). The expression levels of Bcr-Abl and pBcr-Abl in K562 and KU812 cells were decreased with imatinib treatment, but there was no change in that of Bcr-Abl and pBcr-Abl in imatinib-resistant K562R and KU812R cells (Fig. 5C).
To determine whether the cytotoxic effect of co-treatment with deferasirox and imatinib was associated with the induction of apoptosis, we measured the fraction of cells in early apoptosis (annexin V+/PIŌĆō) by flow cytometry. The percentage of annexin V+/PIŌĆō cells did not increase with imatinib or deferasirox single treatment, but the percentage of annexin V+/PIŌĆō cells increased significantly after combined treatment for both imatinib-resistant cell lines (Fig. 6).
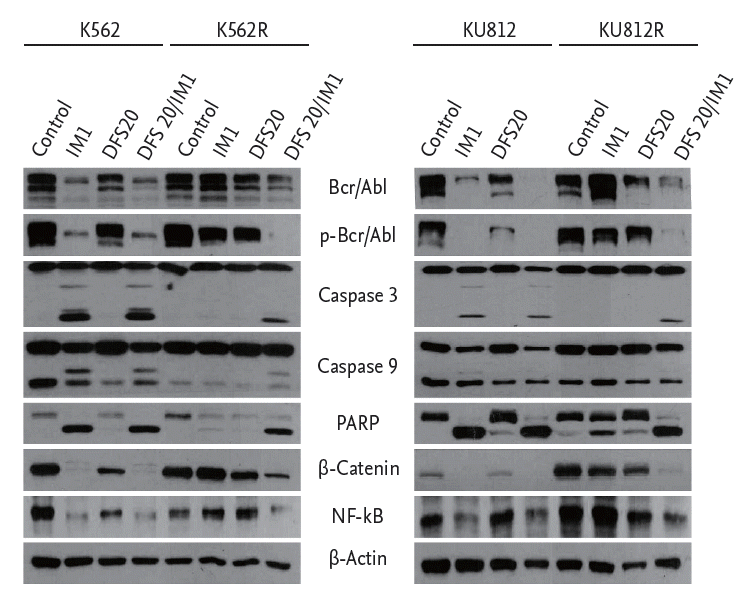
Co-treatment with deferasirox and imatinib significantly down-regulates Bcr-Abl and down-stream pathways of human CML cells
Lastly, to determine the signaling pathways affected by co-treatment with deferasirox and imatinib in imatinib-resistant CML cells, the expression levels of Bcr-Abl and down-stream signaling pathways were evaluated by Western blot analysis. As found with imatinib-sensitive cells, the expression levels of cleaved PARP and cleaved caspase 3/9 were increased after co-treatment in imatinib-resistant CML cells. However, the expression level of cleaved PARP and cleaved caspase 3/9 did not increase with imatinib single treatment in imatinib-resistant cells, in contrast to the results with imatinib-sensitive cells. As shown in Fig. 7, the levels of NF-╬║B and ╬▓-catenin were down-regulated by co-treatment of imatinib-resistant CML cells. However, the Western blot results indicated that imatinib single treatment did not decrease the level of NF-╬║B and ╬▓-catenin in imatinib-resistant CML cells.
DISCUSSION
This study provides an analysis of the synergistic effects of the iron chelator deferasirox and the Bcr-Abl TKI, imatinib on imatinib-resistant CML cells. Co-treatment with deferasirox and imatinib had synergistic effects on not only imatinib-sensitive cells but also imatinib-resistant cells. Co-treatment inhibited cell proliferation and induced accumulation of cells in the subG1 phase. Induction of apoptosis was confirmed by Western blot and flow cytometry analyses. The results of the Western blot assay showed that co-treatment with deferasirox and imatinib reduced the expression levels of Bcr-Abl, pBcr-Abl, NF-╬║B, and ╬▓-catenin in imatinib-resistant cells.
To overcome imatinib resistance, novel ATP-competitive small molecule TKIs, dasatinib and nilotinib, have been developed [9,10]. These drugs have shown efficacy in almost all CML patients. However, some Bcr-Abl mutations such as the T315I mutation, remain resistant to these drugs. In addition, there are other causes of imatinib resistance. Thus, the development of alternative treatments is needed, and it is necessary to identify agents that specifically target the pathways down-stream of the Bcr-Abl signaling pathway to treat CML. Deferasirox is a widely used orally administered iron chelator, which is used to reduce iron overload in patients with hematologic disease. One of the advantages of deferasirox is that it is easy to take and is well tolerated, unlike other iron chelators. Because several studies have reported the antitumor and iron chelating effects of deferasirox against solid tumors and leukemia cells, a synergistic effect is expected when deferasirox is used in combination with other anticancer agents [16,19]. We revealed the antitumor effect of the combined treatment of deferasirox and imatinib in CML cells, and this effect was confirmed to be a synergistic effect by the Chou and Talalay [23] method.
Constitutive NF-╬║B activation has been demonstrated in hematologic malignancies [25,26]. Moreover, NF-╬║B has been identified as a down-stream component of the Bcr-Abl initiated signaling pathway that is necessary for bone marrow cell transformation and leukemogenesis in CML [27]. The role of deferasirox as a NF-╬║B inhibitor has been reported in several previous studies [19,21]. In our study, the expression level of NF-╬║B (p65) was down-regulated by co-treatment with deferasirox and imatinib. This effect was also observed in imatinib-resistant cells. It has been reported that the blockade of NF-╬║B signaling pathway could overcome imatinib-resistance, a finding that is consistent with the results of our study [28-30].
After treatment with imatinib, some residual leukemic cells, most likely leukemic stem cells (LSCs), are resistant to imatinib treatment. The inhibition of Bcr-Abl kinase activity alone is insufficient to eradicate LSCs [31]. The Wnt/╬▓-catenin signaling pathway plays an important role in the survival and self-renewal of imatinib-resistant LSCs and has been linked to many cancers [32]. ╬▓-Catenin is also stabilized by the Bcr-Abl protein and is involved in Bcr-Abl leukemogenesis [33]. Our results showed that the baseline level of ╬▓-catenin in imatinib-resistant CML cells was increased as compared with imatinib-sensitive CML cells. This means that a number of imatinib-resistant CML cells are LSCs. Treatment with deferasirox and imatinib in combination resulted in depletion of Bcr-Abl and pBcr-Abl and down-regulation of ╬▓-catenin, whereas imatinib alone did not affect Bcr-Abl and ╬▓-catenin levels in imatinib-resistant CML cells. These observations indicate that co-treatment can eradicate imatinib-resistant CML cells by depleting Bcr-Abl and ╬▓-catenin, thereby disrupting the Bcr-Abl/╬▓-catenin axis that is essential for LSC survival and self-renewal.
The results of this study are derived from experiments with cell lines in vitro. Animal experiments should be undertaken to confirm these results in vivo. There are still some challenges regarding the clinical applications of deferasirox and imatinib because of side effects, such as gastrointestinal distress; these side effects might be the determining factor for drug dosing and/or compliance. However, it is worth noting that deferasirox showed antileukemic effects against imatinib-resistant CML cells and the synergistic effect occurred with a combination of easily accessible drugs, rather than with cytotoxic chemotherapeutic agents. Based on this study, we suggest that a combination treatment of deferasirox and imatinib may be a suitable alternative treatment option for imatinib-resistant CML.
KEY MESSAGE
1. Co-treatment of deferasirox and imatinib showed anti-leukemic effects against both imatinib-sensitive and imatinib-resistant chronic myeloid leukemia cells.
2. Anti-leukemic effects of deferasirox are due to induction of apoptosis and cell cycle arrest by down-regulated expression of nuclear factor-╬║B and ╬▓-catenin levels.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





