


INTRODUCTION
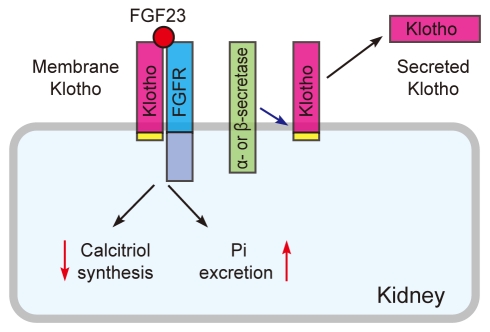
The klotho gene encodes a single-pass transmembrane protein and is expressed primarily in the kidney [1]. A defect in Klotho expression in mice leads to a syndrome resembling aging [1], whereas overexpression of Klotho in mice extends life span [2]. The Klotho protein is composed of a large extracellular domain (- 130 kD), a transmembrane domain, and a very short intracellular domain (10 amino acids). The extracellular domain has homology to family 1 glycosidases (enzymes that hydrolyze terminal glycosidic linkages in sugars, glycoproteins, and glycolipids) and is subject to ectodomain shedding. As a result, the entire extracellular domain is released into the extracellular space and is detectable in blood, urine, and cerebrospinal fluid [2-5]. Thus, the Klotho protein exists in two forms: membrane Klotho and secreted Klotho (Fig. 1). Membrane Klotho functions as a receptor for a hormone that regulates excretion of phosphate and synthesis of active vitamin D in the kidney [6-8]. Secreted Klotho functions as a humoral factor with pleiotropic activities, including suppression of growth factor signaling, suppression of oxidative stress, and regulation of ion channels and transporters [9-12].
Function of membrane Klotho
The function of membrane Klotho was not clear until we realized that Klotho-deficient mice and FGF23-deficient mice suffered identical phenotypes. FGF23 was originally described as one of the 22 members of the fibroblast growth factor (FGF) family [13]. The function of FGF23 became clear when FGF23 was identified as the gene mutated in patients with autosomal dominant hypophosphatemic rickets (ADHR) [14]: ADHR is characterized by hypophosphatemia due to phosphate wasting into urine, inappropriately low serum levels of active vitamin D (1,25-dihydroxy vitamin D3 or calcitriol), and a defect in bone mineralization (rickets). ADHR patients carry missense mutations in the FGF23 gene, which confer resistance to proteolytic inactivation of FGF23 [15]. As a result, serum FGF23 levels are elevated in patients with ADHR. Thus, gain-of-function mutations in the FGF23 gene lead to phosphate wasting, hypophosphatemia, and rickets. These symptoms can be explained by the fact that FGF23 is a bone-derived hormone that acts on the kidney to increase phosphate excretion into urine (phosphaturia) and to suppress calcitriol synthesis [16].
In contrast, FGF23-deficient mice suffer phosphate retention phenotypes, including hyperphosphatemia and vascular calcification [17]. However, in addition to these expected phenotypes, FGF23-deficient mice unexpectedly develop complex phenotypes resembling aging, including growth arrest, kyphosis, osteopenia, emphysematous lung, and atrophy of the gonads, thymus, muscle, skin, and intestine, which are reminiscent of Klotho-deficient mice [18]. Conversely, in addition to aging-like phenotypes, Klotho-deficient mice exhibit hyperphosphatemia and vascular calcification similar to FGF23-deficient mice [1,19,20]. These observations led us to hypothesize that Klotho and FGF23 might function in a common signal transduction pathway.
We found that FGF23 requires membrane Klotho to bind to cognate FGF receptors [6], which was later confirmed independently by other laboratories [7,21]. Although FGF23 belongs to the FGF family, it cannot bind to the FGF receptor (FGFR) with high affinity. Instead, FGF23 uses the Klotho-FGFR complex as its high-affinity receptor: Klotho forms a constitutive binary complex with FGFR1c, FGFR3c, or FGFR4, thereby creating a de novo high-affinity binding site for FGF23 [22]. In other words, Klotho functions as an obligate co-receptor for FGF23. This explains why mice lacking FGF23 (hormone) and Klotho (receptor) develop identical phenotypes. Furthermore, kidney-specific expression of Klotho explains why FGF23 identifies the kidney as its target organ among many other tissues that express multiple FGFR isoforms. The Klotho-FGF23 system may represent a novel mechanism by which a redundant ligand-receptor system secures tissue specificity.
Endocrine regulation of phosphate homeostasis
Phosphate homeostasis is maintained by a counterbalance between absorption of dietary phosphate from the intestine and excretion of blood phosphate from the kidney into urine [23]. These processes are coordinately regulated by several endocrine factors. Calcitriol and parathyroid hormone (PTH), which have been extensively studied as hormones that regulate calcium metabolism, are also involved in phosphate metabolism [24,25]. Calcitriol is secreted by the kidney and acts on the intestine to increase absorption of calcium and phosphate, thereby inducing a positive phosphate balance. PTH is secreted from the parathyroid glands in response to decreases in serum calcium levels and acts on the kidney to promote calcitriol synthesis. However, unlike calcitriol, PTH does not induce a net positive phosphate balance, because PTH promotes phosphate excretion into urine at the same time. Calcitriol in turn suppresses PTH secretion and closes a negative feedback loop. Thus, it had been thought until quite recently that phosphate metabolism was regulated indirectly by calcium-regulating hormones (calcitriol and PTH).
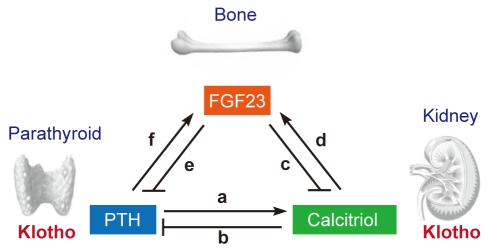
Discovery of the FGF23-Klotho endocrine system has transformed this classic view. One critical feature of FGF23 is that it requires Klotho to be expressed in target organs [11]. FGF23 is secreted from bone and acts on the kidney where Klotho is expressed. FGF23 promotes phosphate excretion and suppresses calcitriol synthesis in the kidney, thereby inducing a negative phosphate balance [16]. Klotho is also expressed in parathyroid glands [26], indicating that parathyroid is another target organ of FGF23. In fact, FGF23 suppresses PTH production and secretion [26], further enhancing its ability to suppress calcitriol synthesis in the kidney. Expression of the FGF23 gene is transactivated with calcitriol in a vitamin D receptor dependent manner [27], forming a negative feedback loop between bone and kidney. Furthermore, PTH increases FGF23 expression [28,29], forming another negative feedback loop between bone and the parathyroid. These newly identified bone-kidney-parathyroid endocrine axes mediated by FGF23 and Klotho add new dimensions to the classic view of endocrine regulation of phosphate homeostasis (Fig. 2, modified from [30,31]).
Phosphate and aging
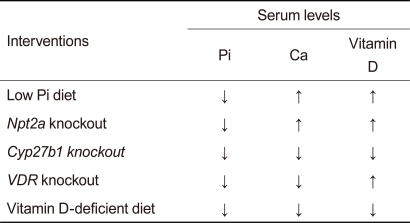
FGF23 functions as a phosphaturic hormone and a counter-regulatory hormone for vitamin D (calcitriol) in a Klotho-dependent manner. Thus, underlying abnormalities caused by defects in the FGF23-Klotho system are hyperphosphatemia and hypervitaminosis D, suggesting that the complex aging-like phenotypes observed in mice lacking FGF23 or Klotho may be attributed to phosphate retention and/or vitamin D intoxication. Several laboratories have successfully rescued the aging-like phenotypes in FGF23-deficient mice and/or Klotho-deficient mice by resolving the hyperphosphatemia and/or hypervitaminosis D by means of dietary and genetic interventions (Table 1, modified from [31]). A vitamin D-deficient diet rescued many aging-like phenotypes of Klotho-deficient mice [19] and FGF23-deficient mice [32]. Similarly, disruption of the vitamin D receptor (VDR) gene [33] or the Cyp27b1 gene (encoding 1a-hydroxylase, an enzyme essential for calcitriol synthesis) [18,34] rescues these mutant mice as well, implying that excess vitamin D activity might be responsible for the aging-like phenotypes. However, these dietary and genetic interventions lowered not only calcitriol but also phosphate and calcium in the blood, raising the possibility that phosphate and/or calcium might be the true culprit (s). In fact, a low phosphate diet reduces blood phosphate levels and rescues mice lacking Klotho [35] or FGF23 [32], despite the observation that it did not reduce blood calcium or calcitriol levels. Similarly, limiting phosphate reabsorption by deleting the kidney-specific sodium-phosphate co-transporter type-2a (Npt2a) gene reduces blood phosphate levels and rescues these mice without restoring serum calcium and calcitriol levels to normal [36]. Thus, it is not calcitriol or calcium but phosphate that is primarily responsible for the aging-like phenotypes caused by defects in the FGF23-Klotho system. These observations indicate that phosphate retention leads to pathologies resembling aging, which may be collectively referred to as "phosphatopathies."
Aging and chronic kidney disease
Phosphatopathies are universally observed in patients with chronic kidney disease (CKD). CKD is defined as a state of progressive decline of renal function over months/years caused by any chronic diseases that affect the kidney (most notably hypertension and diabetes) and/or natural aging. More than 26 million Americans, or 13% of the total population, have CKD, which is increasingly recognized as a public health problem in the aging society [37,38]. Since hyperphosphatemia was identified as a potent mortality risk in CKD patients [39-41], lowering blood phosphate levels below 4.5 mg/dL with a low phosphate diet and phosphate binders (medications that chelate phosphate in the gut and prevent its absorption) has been an important therapeutic goal in the management of patients with CKD [42].
Patients with CKD exhibit a marked decrease in renal Klotho expression associated with resistance to FGF23, hyperphosphatemia, and vascular calcification, which are reminiscent of Klotho-deficient mice [43-45]. Notably, the vast majority of patients with CKD die prematurely not due to renal failure, but due to early onset of common age-related diseases such as cardiovascular disease, cancer, and infection [46,47]. Consequently, the spectrum of death causes in patients with CKD is similar to that of the general population. Patients with CKD also suffer many aging-like symptoms, including hypogonadism, skin atrophy, osteopenia, and cognitive impairment. Thus, CKD may be viewed as a state of accelerated aging and/or an age-related disease associated with Klotho deficiency and phosphate retention.
Phosphate metabolism in CKD
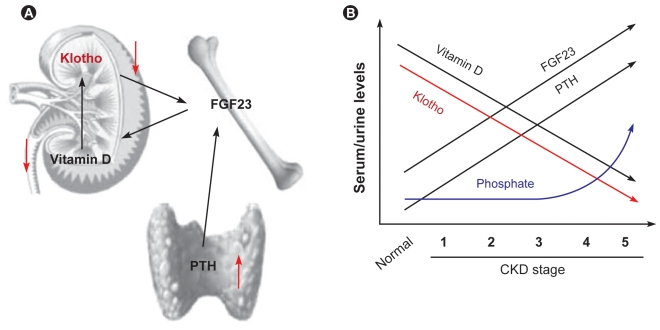
As functional nephrons are progressively lost during the progression of CKD, each nephron is required to excrete an increasing amount of phosphate to prevent phosphate retention. This can be achieved by increasing serum levels of FGF23. However, FGF23 suppresses calcitriol synthesis in the kidney at the same time. Decreases in functional nephrons capable of calcitriol synthesis may also contribute to decreases in calcitriol. In fact, increases in FGF23 and decreases in calcitriol precede overt hyperphosphatemia during the progression of CKD [41]. These observations indicate that early-stage patients with CKD maintain normal blood phosphate levels by increasing FGF23 to compensate for the reduced ability of the kidney to excrete phosphate at the expense of calcitriol. Because calcitriol is a potent upregulator of Klotho [19], decreases in calcitriol can reduce Klotho expression. In fact, decreases in urine Klotho levels are observed in the early-stage of CKD long before hyperphosphatemia ensues [48]. Because calcitriol also functions as a potent suppressor of PTH [24], decreases in calcitriol can increase PTH. In fact, serum PTH levels start increasing during the early-stage of CKD prior to the start of the increase in serum phosphate levels [41]. Decreases in Klotho can induce FGF23 resistance in the kidney and parathyroid. Moreover, increases in PTH can increase FGF23 [29]. Thus, these changes in phosphate-regulating hormones and Klotho form a vicious cycle, leading to high FGF23, high PTH, low calcitriol, and low Klotho in patients with end-stage renal disease (Fig. 3, modified from [30,31]). Hyperphosphatemia ensues when the number of functional nephrons decreases to a level that fails to excrete ingested phosphate into urine.
Notably, phosphatopathies such as vascular calcification can occur in patients with CKD and normal blood phosphate levels, which is associated with an increased incidence of cardiovascular events [46,49,50]. One possible explanation for this dissociation would be that these patients might suffer postprandial hyperphosphatemia, which may be sufficient to induce phosphatopathies even if they are normophoshatemic under fasting conditions. Because dietary phosphate overload can increase blood phosphate levels transiently, even in healthy individuals [51], it is possible that patients with CKD and a reduced ability to excrete phosphate into urine may suffer prolonged and/or enhanced postprandial hyperphosphatemia. Another possible explanation would be that tissue phosphate content might be increased in patients with CKD even if their serum phosphate levels are normal. Secreted Klotho protein has activity that suppresses phosphate uptake in various types of cells, including vascular smooth muscle cells, by inhibiting type-III sodium-dependent phosphate co-transporters, which may protect arteries from calcification [48]. Decreased Klotho expression in the kidney of patients with CKD potentially reduces blood levels of secreted Klotho and promotes vascular calcification.
Function of secreted Klotho
Membrane Klotho is clipped on the cell surface by membrane-anchored proteases (α- and β-secretases), resulting in the secretion of the entire extracellular domain (- 130 kD) into systemic circulation [3-5]. Secreted Klotho has pleiotropic functions as a humoral factor independently of FGF23 (Table 2).
Secreted Klotho belongs to family 1 glycosideses and has weak glycosidase activity in vitro [52]. However, endogenous substrates for secreted Klotho were not clear until the mechanism by which secreted Klotho activates the TRPV5 calcium channel was revealed [53,54]. TRPV5 is expressed on the luminal side of distal tubules in the kidney and functions as the rate-limiting entry gate for transepithelial calcium reabsorption [55]. TRPV5 activity depends on the number of channels present on the cell surface, which is regulated by the rate of endocytosis. Secreted Klotho suppresses TRPV5 endocytosis by modifying its N-linked glycans and increasing the cell-surface abundance of TRPV5; secreted Klotho removes terminal sialic acids in the N-glycan of TRPV5 on the cell surface through its putative sialidase activity. Removal of terminal sialic acids by secreted Klotho exposes underlying galactose residues in the N-glycan, which are ligands for a ubiquitous galactose-binding lectin called galectin-1. Binding of the modified N-glycan to the galectin-1 lattice in the extracellular matrix suppresses TRPV5 endocytosis, leading to the accumulation of TRPV5 on the cell surface [54]. The ROMK1 potassium channel is also activated by secreted Klotho through the same mechanism [56].
The putative sialidase activity of secreted Klotho may also be involved in its ability to suppress kidney-specific sodium-dependent phosphate co-transporter type-IIa (Npt2a) [57]. Npt2a is expressed on the brush border membrane of renal proximal tubular cells and functions as the major entry gate for transepithelial phosphate reabsorption. Modification of the N-glycan of Npt2a by secreted Klotho leads to inactivation and proteolytic degradation of Npt2a through a mechanism yet to be identified. Secreted Klotho also suppresses type-III sodium-dependent phosphate co-transporters (Pit-1 and Pit-2), which are ubiquitously expressed to mediate cellular phosphate uptake [48]. It is possible that the ability of secreted Klotho to suppress these sodium-dependent phosphate co-transporters may contribute to preventing phosphatopathies.
Secreted Klotho regulates the activity of multiple growth factors, including insulin/insulin-like growth factor-1 (IGF-1) [2], Wnt [58], and transforming growth factor (TGF)-β1 [59]. Because adequate suppression of insulin/IGF-1 signaling pathway has been identified as an evolutionarily conserved mechanism for extending life span [60], the anti-aging properties of Klotho may stem partly from its ability to suppress insulin/IGF-1 signaling. In fact, transgenic mice that overexpress Klotho are long-lived and slightly resistant to insulin and IGF-1 without overt diabetes [2]. The mechanism by which secreted Klotho suppresses insulin/IGF-1 signaling remains to be determined. However, secreted Klotho inhibits Wnt signaling by directly binding to Wnt ligands and preventing them from binding to their receptors. Wnt signaling is enhanced in Klotho-deficient mice, which results in exhaustion of stem cells in highly proliferative tissues such as skin and intestine and may partly contribute to atrophy of these tissues in Klotho-deficient mice [58].
We recently found that secreted Klotho suppresses TGF-β1 signaling; secreted Klotho directly binds to type-II TGF-β receptor (TGFβR2) on the cell surface and prevents TGF-β1 binding to TGFβR2 [59]. TGF-β1 is the most potent inducer of the epithelial-to-mesenchymal transition (EMT) [61]. EMT is a cellular process whereby epithelial cells lose epithelial characters and undergo a phenotypic transition to acquire mesenchymal characters, including the ability to migrate and proliferate. EMT is essential for tissue repair in response to injury but can result in fibrosis under pathological conditions in the kidney as well as in many other tissues, including the liver, lung, and heart [62]. Furthermore, cancer cells undergo EMT and acquire the ability to migrate and proliferate, leading to metastasis [62]. Thus, the ability of secreted Klotho to inhibit TGF-β1 activity may counteract EMT and prevent tissue fibrosis and cancer metastasis. In fact, injecting secreted Klotho prevents renal fibrosis induced by unilateral ureteral obstruction and metastasis of human cancer xenografts in mice [59]. These activities of secreted Klotho may also contribute to life span extension by Klotho overexpression in mice [2].
The Klotho family and endocrine FGFs
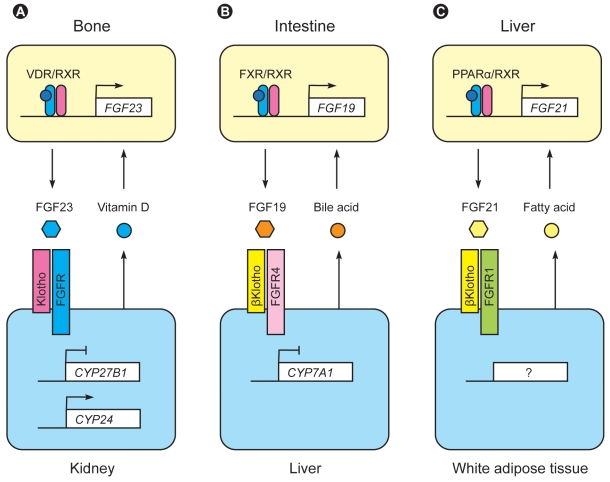
After the discovery of the klotho gene, two Klotho-related genes were identified based on sequence similarity and designated as βKlotho [63] and γKlotho (also known as KLPH or LCTL) [64]. The original Klotho has been referred to αKlotho when necessary to distinguish it from βKlotho and γKlotho. Similar to αKlotho, βKlotho and γKlotho are single-pass transmembrane proteins that form complexes with FGF receptors [65-67]. βKlotho is expressed in various tissues, most notably in the liver and white adipose tissue, and forms complexes with FGFR1c and FGFR4 [66-68]. γKlotho is expressed in eyes and forms complexes with FGFR1b, FGFR1c, FGFR2c, and FGFR4 [67]. Importantly, these Klotho-FGFR complexes function as high-affinity receptor for endocrine FGFs other than FGF23. A phylogenetic analysis has segregated FGF23 and two additional FGFs (FGF19 and FGF21) from the other FGF family members. These three FGFs, FGF19, FGF21, and FGF23, are collectively called endocrine FGFs, because they function as hormones, unlike the other FGFs that function primarily as paracrine/autocrine factors [69]. One critical feature of endocrine FGFs is that they require Klothos to be expressed in their target tissues and exert their biological activity. FGF19 requires βKlotho [66] or γKlotho [67], whereas FGF21 requires βKlotho [65].
FGF19 is secreted from the intestine upon feeding and acts on the liver to suppress bile acid synthesis by downregulating Cyp7a1 gene expression, which encodes the bile acid synthesis rate-limiting enzyme [70]. This intestine-liver endocrine axis mediated by FGF19 and βKlotho is indispensable for maintaining bile acid homeostasis, because mice lacking either FGF15 (the mouse ortholog of human FGF19), βKlotho, or FGFR4 (the prominent FGFR isoform expressed in the liver) exhibit increased Cyp7a1 expression and bile acid synthesis in the liver [70-72]. Bile acids bind to the FXR nuclear receptor and transactivate the FGF19 gene in the intestine, forming a negative feedback loop between the intestine and liver [70] (Fig. 4, modified from [69]).
In contrast to FGF19, FGF21 is secreted from the liver upon fasting and acts on white adipose tissue to induce lipolysis and the metabolic adaptation to fasting [73]. Fatty acids bind to the PPARα nuclear receptor and transactivate the FGF21 gene in the liver, forming a negative feedback loop between white adipose tissue and liver. Of note, transgenic mice that overexpress FGF21 are subject to torpor, a short-term hibernation-like state whereby animals reduce activity and body temperature to limit energy expenditure [73]. These mice are smaller than wild-type mice due to resistance to growth hormone and low serum IGF-1 levels [74], suggesting that FGF21 may extend life span by suppressing the somatotroph-endocrine axis.
These observations indicate that the most important function of the Klotho family (αKlotho, βKlotho, and γKlotho) is to regulate the function of endocrine FGFs (FGF19, FGF21, and FGF23) [69]. These newly identified endocrine axes mediated by endocrine FGFs and Klothos regulate various metabolic processes and have a common negative feedback structure composed of lipophilic ligands (bile acid, fatty acid, and vitamin D), nuclear receptors (FXR, PPARα, and VDR), and cytochrome P450 superfamily enzymes (Cyp7a1 and Cyp27b1) (Fig. 4).
CONCLUSIONS
Since the discovery of the klotho gene in 1997 [1], we have witnessed several breakthroughs that have significantly advanced our understanding of Klotho protein function. Membrane Klotho functions as the obligate co-receptor for FGF23, a bone-derived phosphaturic hormone. Defects in either FGF23 or Klotho result in phosphate retention associated with complex aging-like phenotypes, which we propose calling phosphatopathies. These findings have revealed an unexpected link between phosphate and aging [75]. Phosphate retention associated with Klotho deficiency is universally observed in patients with CKD, suggesting that CKD may be viewed as a state of accelerated aging. Discovery of the Klotho family members have identified multiple novel endocrine axes and opened a novel research field in endocrinology. Further studies on the Klotho family are expected to provide new insights into endocrine regulation of various metabolic and aging processes.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





