


INTRODUCTION
Hutchinson-Gilford progeria syndrome (HGPS) and Werner's syndrome are two well-characterized disorders that present with most of the features of premature aging. HGPS is a recent member of the family of "laminopathies", and these are caused by mutations in LMNA which are inherited either autosomal dominant (AD) or autosomal recessive (AR) manner. Most HGPS probands are heterozygous for LMNA G608G, but one atypical HGPS proband was found to have an E145K mutation and one recently reported patient was a compound heterozygote for R471C and R527C mutations. Furthermore, a small subset of patients with the features of the adult-onset premature aging Werner's syndrome (WRN, OMIM 277700), but without mutations in the causative WRN/RECQL2 gene was found to have novel heterozygous LMNA mutations [1]. Werner's syndrome is an autosomal recessive disorder and this is caused by the Werner syndrome gene (WRN), which encodes a RecQ-type DNA/RNA helicase [2]. The gene is located on the 8p11-12 locus and it encodes a 1432 amino acid protein (WRN) that's homologous to the RecQ subfamily helicases [1].
Children with HGPS usually appear normal at birth, but they experience profound failure to thrive during the first year. Characteristic facies, alopecia, lose of subcutaneous fat, stiffness of joints, bone changes and abnormal tightness of the skin over the abdomen and upper thighs usually become apparent during the second or third year [3]. On the other hand, patients with Werner's syndrome have a bird-like appearance of the face [4]. The clinical manifestations of Werner's syndrome occur during the third or fourth decade, and these patients die prematurely during the fourth or fifth decade [4,5]. HGPS is a rare, autosomal dominant disorder and it is caused by specific synonymous heterozygous de novo mutations that affect the codon 608 of the LMNA gene, which normally encodes for a glycine residue [6-9]. These mutations generate an alternative splicing site in exon 11, resulting in a truncated lamin A protein.
The other form of progeroid syndrome, atypical Werner's syndrome, was first reported on in 2003 by Lishan Chen, etc [10]. These patients have heterozygous missense mutations in the LMNA gene, which were distinct from those reported in patients with typical HGPS. We found atypical Werner's syndrome in a 15-yr-old woman with lipodystrophy, steatohepatitis, type 2 diabetes mellitus and progeroid features, and the patient had T156del in the LMNA gene.
CASE REPORT
Clinical and laboratory history
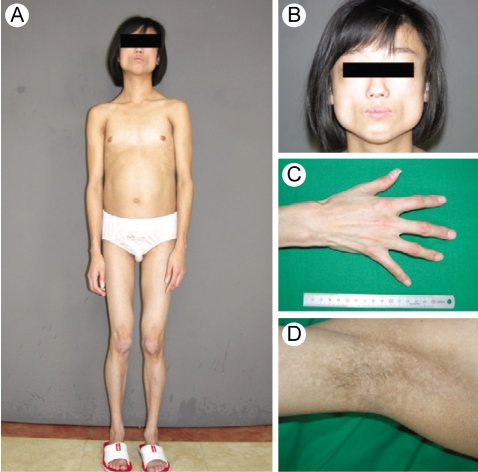
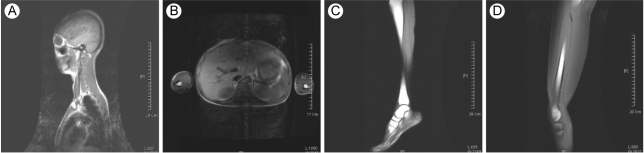
A 15-yr-old woman presented with abdominal distension and an elevated blood glucose level. She was born at full term with a birth weight of 2.7 kg by c-section delivery. Hyperpigmentation of her skin developed 10 months from birth. She attained menarche at the age of 13 yr, and this was followed by an irregular period. She developed diabetes mellitus at the age of 13 yr and began diabetic treatment with oral hyperglycemic agents (sulfonylurea, metformin, voglibose). Yet she didn't achieve good glycemic control despite using oral hypoglycemic agents. On admission, her random plasma glucose was measured by the glucose oxidase method using a Hitachi Modular D2400 (Roche, Tokyo, Japan) and the serum cholesterol, triglycerides, high-density lipoprotein cholesterol and chemistry were tested using a Hitachi Modular D2400 (Roche, Tokyo, Japan). Her fasting plasma glucose (13.2 mmol/liter), triglyceride (14.9 mmol/liter), alanine aminotransferase (87 U/L), and aspartate aminotransferase levels (70 U/L) were all increased. She had severe hepatomegaly on the MR imaging. Microvesicular and macrovesicular changes and perivenular fibrosis were noted on the liver biopsy. On examination, she looked older than her age group and she had the bird-like face with a beaked nose (Fig. 1A and 1B). The skin over the hands and feet was atrophic with prominent superficial veins (Fig. 1C). She had thin, spindle-shaped fingers with mild flexion contractures of the distal and proximal interphalangeal joints and elbow joints (Fig. 1C). She had a small mandible and a small mouth (Fig. 1B). There was hyperpigmented skin covering the neck, trunk and upper extremities (Fig. 1D). She had generalized loss of subcutaneous fat over the face, neck, upper chest area, hips, and extremities (Fig. 1A and 1C). The lateral quadriceps were prominent bilaterally. There was no graying, but there was thinning of scalp hair and diminution of hair over her eyebrows and over her upper and lower extremities. The liver was palpable 5 cm below the right costal margin, and the spleen was not palpable. She didn't have cataract or any abnormality in fundus exam. Doppler echocardiography did not show concentric left ventricular hypertrophy, increased left ventricular pressures, aortic or mitral regurgitation, or calcification of the posterior annulus. The whole-body fat and regional fat in the head, trunk and upper and lower extremities were determined by performing dual-energy X-ray absorptiometry (DEXA) scanning (GE Lunar [Madison, WI] instruments). The proportion of fat in the individual lesions as well as that of the whole body was calculated as a percentage of the body mass. She had markedly reduced body fat with generalized lipodystrophy. The body fat with using DEXA was estimated to be 6.5% of the total body mass. Whole body MRI studies confirmed markedly decreased subcutaneous fat, abdominal fat and intrathoracic fat (Fig. 2). MRI studies were performed using a 3 Tesla imaging device (GE Signa Exite 3.0 HD T) at the trunk (chest, abdomen) and the peripheral (biceps, triceps, forearm, thigh, and calf) limbs. Her grandmother was diagnosed with diabetes mellitus at 72 yr old, but her parents, brother and sister did not have any history of diabetes or cardiovascular disease. Her parents, brother, and younger sister didn't have an abnormal appearance of their faces, extremities, skin and etc. We did not find abnormal finding in their medical histories.
Lamin A/C immunofluorescence in skin fibroblasts
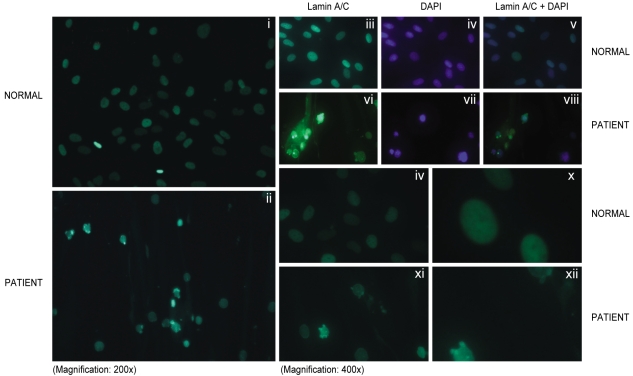
Primary fibroblasts from skin biopsy samples were grown on Dulbecco's modified Eagle's medium (DMEM, Gibco Lab, NY, USA) and they were fixed in phosphate buffered saline (PBS) that contained 4% formaldehyde for 30 minutes at 4℃. The cells were made permeable by incubating them in 0.1% Triton X-100 for 15 min at room temperature and then they were blocked for nonspecific binding by incubation with monoclonal anti-lamin antibody (antibody H-110 at a 1:100 dilution in blocking buffer; Santa Cruz Biotechnology, Santa Cruz, CA.) for 60 min at 37℃. The primary antibody was removed, and the coverslip was washed with PBS and then incubated with the secondary antibody conjugated with FITC-conjugated goat anti-rabbit IgG antibody for 120 min at room temperature. After washing, the coverslips were mounted using commercial mounting medium for fluorescent microscopy (Aqua Poly/ Mount; Polysciences, Inc., Warrington, PA.), and the slides were examined using an AxioCam MRc5 microscope (Carl Zeiss USA, Thornwood, NY). The skin fibroblasts from this patient were from the 7th passage. Indirect immunofluorescent studies revealed normal localization of the lamin A/C protein in the nuclear envelope in the affected subject. The captured images of the nuclei showed abnormal nuclear morphology. Several deformed nuclei, multilobulations, nuclear membrane invagination and nuclear hypertrophy were observed in this patient (Fig. 3).
Exon 2 sequence of the LMNA DNA
We found the exonic sequence associated with the T506del and it was 25.374 Kb of genomic DNA. The nucleotide numbering started with the adenine of the first ATG codon for lamin A/C. We found a T deletion in exon 2 in the LMNA gene that resulted in altered amino acid sequence from codon 169 to stop codon (Fig. 4). Although G507del was found in LMNA gene of her mother and sister, they didn't have any phenotype of premature aging or lipodystrophy. The exonic sequence of the LMNA gene of her father and brother had the same sequence compared with other normal persons. The exonic sequence of the LMNA gene of each person is shown in Figure 4.
DISCUSSION
Progeroid syndrome is a rare disorder that is caused by gene mutation. Progeroid syndrome is divided into 5 subtypes by the mutation site of the gene and the phenotypes [1]. The common types among them are HGPS and Werner's syndrome. Atypical Werner's syndrome was recently reported on [6,10]. The patients with atypical Werner's syndrome have some different phenotypes compared to the phenotypes of patients with Werner's syndrome.
For the atypical Werner's syndrome that was reported in 2003 by Lishan Chen, the patients had heterozygous R133L, L140R and A57P LMNA mutations [10]. In 2005, two patients were evaluated at the General Clinical Research Center of the University of Texas Southwestern Medical Center at Dallas. It was detected that the patients had A133L due to being heterozygous for the 398G>T LMNA mutation [6]. Phenotypic variation has been observed in other patients that suffer from laminopathies [6,11,12]. The patients with atypical Werner's syndrome have mutation of the LMNA gene that codes for the intermediate filament lamin, and so they have various phenotypes such as myopathy, lipodystrophy, mandibuloacral malformation and etc.
The hypotheses about how LMNAgene mutation induces the disease include alteration of nuclear structure, cell instability and tissue atrophy [10,13]. For the second hypothesis, the intermediate filament lamin acts to regulate several important nuclear components whose activities are important to prevent disease onset [10,14]. Yet there is no information about the pathophysiology of tissue specificity [10].
A hypothesis on how the progeroid features in atypical Werner's syndrome develop has been suggested. The hypothesis was that abnormal function of retinoblastoma protein may be associated with the progeroid phenotypes [10]. In this case, we didn't check if she had abnormal function of retinoblastoma protein, but we observed the abnormal shapes of the fibroblast nuclei.
T506del in LMNA has not been previously reported on in a patient with atypical Werner's syndrome. We found that this patient had the T506del and the early expression of the stop codon. We investigated for genetic mutation of her family members. Her father and younger brother had normal genetic arrangement compared with the normal control group. Her mother and younger sister had the G507del, resulting in a change of amino acid arrangement compared to normal populations. The amino acid arrangement of our patient was the same as her mother and younger sister, except for a valine substitution for glycine. But her mother and younger sister did not show abnormal findings in their appearance. The T506del results in disease, but the G507del didn't induce abnormal phenotypes in her mother and sister, despite the abnormal arrangement in the amino acid. We didn't investigate the WRN gene mutation in this patient and her family members because her external features and clinical symptoms were similar to those of the reported patients with atypical Werner's syndrome [6].
In summary, we found a new patient with atypical Werner's syndrome, and this patient had mutation of the T506del in the LMNA gene. She had characteristic progeroid features, generalized lipodystrophy, diabetes mellitus, hypertriglyceridemia and a fatty liver. We found that the cause of disease was deletion of the LMNA gene. However, the molecular mechanism of the T506del in the LMNA gene, and the relationship between the other mutations in the LMNA gene and the clinical heterogeneity both remain to be determined.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





